
The Female Hormone Pathway: Estrogen, Progesterone, And The Cycling System That Governs Reproductive Health
By Jacob Gordon, INHC, FMT-CFemale hormone physiology is not a single hormone level.
It is a dynamic system, a precisely timed orchestration of two cell types in the ovary, two gonadotropins from the pituitary, a feedback loop that switches from negative to positive partway through each cycle, a corpus luteum that lives and dies on a predictable schedule, and a cascade of downstream effects on every tissue from bone to brain to vasculature.
Understanding this system means understanding why PCOS is fundamentally a problem of LH excess and theca cell hyperactivation, why perimenopause is a phase of erratic estrogen rather than simple decline, why progesterone is not just a pregnancy hormone, and why the two estrogen receptors have nearly opposite effects in some tissues.
The Three Estrogens And What They Do
There are three biologically significant estrogens: R
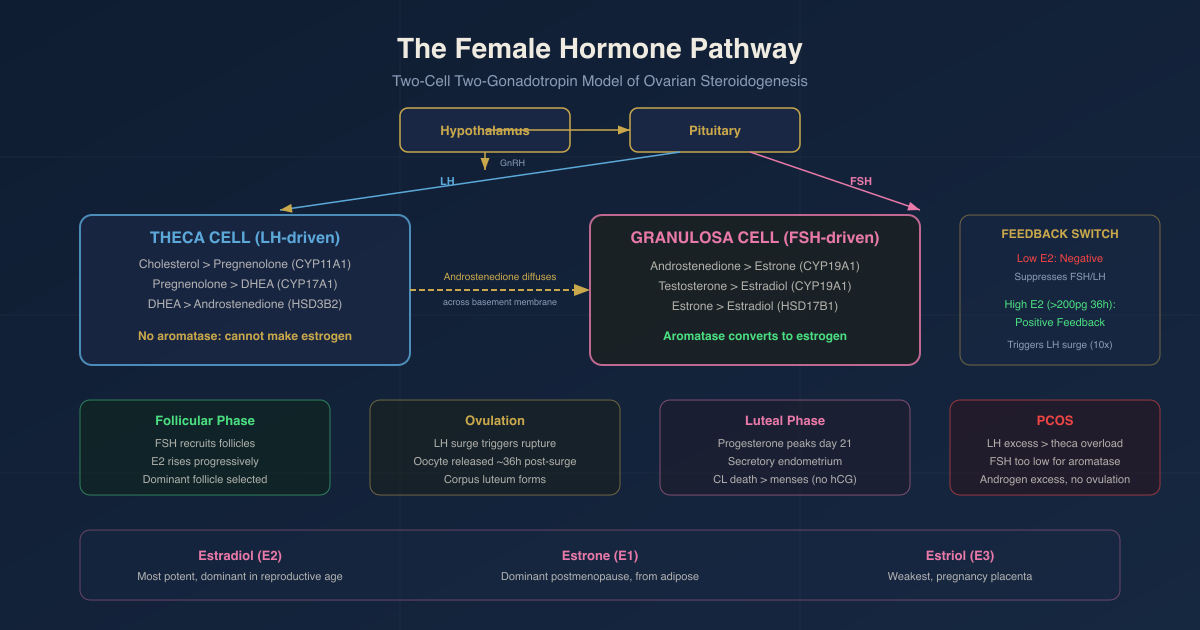
Estradiol (E2): the most potent and the dominant estrogen in reproductive-age women.
E2 is produced primarily by granulosa cells in the ovary during the follicular phase and by the corpus luteum during the luteal phase.
Normal range varies dramatically by cycle phase: approximately 30-400 pg/mL during the follicular phase, peaking at 200-500 pg/mL just before ovulation, then 100-300 pg/mL in the luteal phase.
E2 drives endometrial proliferation, bone formation, vaginal lubrication, favorable lipid profiles, serotonin modulation, and the primary negative feedback signal to the pituitary that controls FSH and LH.
At the preovulatory surge, E2 switches from negative to positive feedback on the pituitary, triggering the LH surge that causes ovulation.
Estrone (E1): a weaker estrogen, primarily the dominant estrogen in postmenopausal women.
E1 is produced by peripheral conversion of androstenedione (primarily from the adrenal gland) by aromatase in adipose tissue.
E1 can be interconverted with E2 by 17beta-hydroxysteroid dehydrogenase type 1 (HSD17B1) in target tissues.
Estriol (E3): produced in large quantities by the placenta during pregnancy from 16alpha-hydroxyDHEA-S derived from the fetal adrenal.
E3 is the weakest of the three estrogens at estrogen receptors.
It is used as a measure of fetal and placental wellbeing during pregnancy.
E3 is also the primary estrogen used in vaginal creams for genitourinary syndrome of menopause (the application most appropriate to its lower potency and local action).
Progesterone And Its Metabolites
Progesterone is a 21-carbon steroid (progestogen) produced primarily by the corpus luteum in the second half of the menstrual cycle, with minor contributions from the adrenal cortex.
Normal range: approximately 0.1-0.3 ng/mL in the follicular phase, rising to 5-20 ng/mL in the mid-luteal phase.
Progesterone's primary functions:
- Converts the proliferative endometrium (built by estrogen) to a secretory state capable of receiving an embryo
- Inhibits further follicular development during the luteal phase (prevents double ovulation)
- Elevates basal body temperature by approximately 0.3-0.5 degrees Celsius (the basis for basal body temperature charting of ovulation)
- Provides cervical mucus changes (from thin, spinnbarkeit mucus favorable to sperm during the follicular phase, to thick, hostile mucus post-ovulation)
- Has immunomodulatory effects supporting early pregnancy
- Acts as a precursor to neurosteroids including allopregnanolone (3alpha,5alpha-tetrahydroprogesterone), a potent positive allosteric modulator of GABA-A receptors with anxiolytic and sedating properties
- Provides partial negative feedback on the HPG axis (opposing estrogen's positive feedback and suppressing LH pulse frequency in the luteal phase)
Allopregnanolone is the reduced metabolite of progesterone (progesterone to 5alpha-dihydroprogesterone to allopregnanolone) produced in the brain, adrenal, and corpus luteum.
Allopregnanolone's GABA-A-potentiating effects are relevant to premenstrual dysphoric disorder (PMDD), in which an abnormal central response to normal allopregnanolone fluctuations occurs, and to brexanolone (Zulresso), the FDA-approved IV allopregnanolone analog for postpartum depression.
17-Hydroxyprogesterone (17-OHP): a progesterone metabolite elevated in congenital adrenal hyperplasia (21-hydroxylase deficiency); measured in newborn screening and in the evaluation of suspected late-onset CAH in women with hyperandrogenism.
The Female HPG Axis
The female hypothalamic-pituitary-gonadal axis operates on the same basic architecture as the male, but with a critical difference: the feedback is not simply negative.
In certain phases of the cycle, estradiol switches from suppressing gonadotropin release to actively stimulating it.
Hypothalamic GnRH:
GnRH neurons in the arcuate nucleus release GnRH in pulses.
GnRH pulse frequency is modulated by kisspeptin neurons in the arcuate nucleus (KNDy neurons, which co-release kisspeptin, neurokinin B, and dynorphin).
Neurokinin B stimulates GnRH pulse initiation; dynorphin slows pulse frequency; kisspeptin directly stimulates GnRH neurons.
In the follicular phase, slower-frequency GnRH pulses favor FSH secretion.
As the follicular phase progresses and estradiol rises, GnRH pulse frequency increases, shifting the pituitary toward LH secretion.
Pituitary FSH and LH:
Follicle-stimulating hormone (FSH) drives follicular recruitment and maturation, and specifically stimulates aromatase expression in granulosa cells.
Luteinizing hormone (LH) stimulates androgen production in theca cells (the substrate for estrogen synthesis) and triggers the final maturation and release of the dominant follicle.
The ratio of FSH to LH matters enormously for follicular health.
High LH relative to FSH (as in PCOS) drives excess androgen production from theca cells faster than granulosa aromatase can convert it to estrogen.
The Two-Cell Two-Gonadotropin Theory Of Ovarian Steroidogenesis
Estradiol synthesis in the ovary cannot be accomplished by any single cell type acting alone.
It requires collaboration between two cell types, each receiving a distinct gonadotropin signal: the two-cell, two-gonadotropin hypothesis. R
Theca cells (LH-responsive):
Theca cells surround the outer layer of the developing follicle.
They express LH receptors and respond to LH by activating adenylyl cyclase (via Gs-coupled receptor), raising cAMP, and activating PKA.
PKA-mediated phosphorylation induces StAR expression, activating the cholesterol delivery to CYP11A1.
CYP17A1 with its 17,20-lyase activity (enabled by cytochrome b5 in theca cells) converts pregnenolone to DHEA, then androstenedione.
Theca cells do not express aromatase (CYP19A1): they cannot complete the conversion to estrogen.
They produce androstenedione and some testosterone, which diffuse across the basement membrane into granulosa cells.
Granulosa cells (FSH-responsive):
Granulosa cells line the follicle interior.
They express FSH receptors and respond to FSH by activating the same cAMP/PKA pathway.
FSH drives the expression of CYP19A1 (aromatase) in granulosa cells via PKA/CREB-mediated transcription and via SF-1 (steroidogenic factor 1) activation.
Granulosa cell aromatase converts theca-derived androstenedione to estrone (E1) and testosterone to estradiol (E2).
17beta-HSD type 1 (HSD17B1) in granulosa cells preferentially converts estrone to the more potent estradiol.
Neither cell type alone can make estradiol efficiently.
Theca cells lack aromatase.
Granulosa cells in small follicles lack the full complement of upstream steroidogenic enzymes needed to make sufficient androgen precursors independently.
The two-cell system is the ovary's solution: LH drives androgen substrate production in theca cells, FSH drives aromatase expression and estrogen production in granulosa cells, and the two outputs are combined across the basement membrane. R
Cholesterol To Estradiol: The Complete Ovarian Pathway
The complete enzymatic sequence in the ovary:
In theca cells (LH-driven):
- Cholesterol to Pregnenolone: CYP11A1 (StAR-dependent, mitochondrial)
- Pregnenolone to 17alpha-hydroxypregnenolone: CYP17A1 (17alpha-hydroxylase activity, ER)
- 17alpha-hydroxypregnenolone to DHEA: CYP17A1 (17,20-lyase activity, requires cytochrome b5)
- Pregnenolone to Progesterone: HSD3B2 (delta-5 to delta-4 conversion)
- Progesterone to 17alpha-hydroxyprogesterone: CYP17A1
- DHEA to Androstenedione: HSD3B2
- Androstenedione is secreted and diffuses into granulosa cells
In granulosa cells (FSH-driven):
- Androstenedione to Estrone (E1): CYP19A1 (aromatase)
- Testosterone to Estradiol (E2): CYP19A1 (aromatase)
- Estrone to Estradiol: HSD17B1 (interconversion, favors E2 in granulosa cells)
Estradiol is the primary secretory product from the follicle during the follicular phase.
After the LH surge, granulosa cells luteinize:
The LH surge dramatically upregulates HSD3B2 and StAR in granulosa cells.
CYP19A1 (aromatase) is downregulated in luteinizing granulosa cells.
The luteinized granulosa cells (now the large luteal cells of the corpus luteum) shift from estrogen production to progesterone production.
This luteal phase shift is the biochemical basis for the biphasic hormone pattern of the menstrual cycle.
The Menstrual Cycle: Phase By Phase
Days 1-5: Menstruation
Estradiol and progesterone have dropped from the prior corpus luteum's demise.
The endometrium sheds.
FSH begins to rise (because low estradiol removes negative feedback from the pituitary).
Rising FSH recruits a new cohort of follicles.
Days 1-13: Follicular Phase
FSH stimulates a cohort of 5-15 antral follicles to grow.
The dominant follicle emerges (usually by day 7-8) due to its higher FSH receptor expression and greater IGF-1 sensitivity.
Growing granulosa cells in the dominant follicle aromatize theca-derived androstenedione to estradiol.
Estradiol rises progressively throughout the follicular phase.
Rising estradiol provides negative feedback to the pituitary: FSH levels decrease, while LH levels are maintained or slightly elevated.
The dominant follicle, with its high aromatase capacity, can survive on lower FSH levels that cause smaller follicles to undergo atresia.
This is follicle selection.
The dominant follicle also produces inhibin B, which selectively suppresses FSH at the pituitary, further reducing FSH levels and preventing other follicles from maturing to compete.
Day 12-14: The Estradiol Surge And Positive Feedback
When E2 levels exceed approximately 200 pg/mL for at least 36-48 hours, a remarkable switch occurs: R
Estradiol switches from negative to positive feedback at the pituitary and hypothalamus.
GnRH pulse frequency increases.
The pituitary releases a massive LH surge (10-fold increase in LH, peak approximately 28 IU/L).
FSH surges too, though less dramatically.
The trigger for positive feedback requires sustained high E2 acting through ERalpha on GnRH neurons (ERE-dependent ERalpha signaling), as well as on pituitary gonadotrophs directly.
This is one of the clearest examples in endocrinology of the same hormone producing diametrically opposite regulatory effects depending on dose, duration, and context.
The LH Surge And Ovulation
The LH surge triggers a cascade of events in the dominant follicle:
Resumption of meiosis: the oocyte, which has been arrested at prophase I since fetal development, resumes meiosis after the LH surge and completes meiosis I, becoming a secondary oocyte arrested at metaphase II.
Cumulus cell expansion: LH induces cumulus cells to secrete hyaluronic acid, forming the cumulus-oocyte complex.
Follicle wall proteolysis: LH stimulates prostaglandin production and matrix metalloproteinase activation in the follicle wall, enabling physical rupture.
Follicle rupture and oocyte release: approximately 36 hours after the LH surge peak.
Corpus luteum formation: the granulosa and theca cells left behind undergo luteinization, forming the corpus luteum under ongoing LH stimulation.
The Corpus Luteum: Progesterone Production And Luteolysis
The corpus luteum (CL) is a transient endocrine gland formed from the ruptured follicle. R
It produces high concentrations of progesterone (and some estradiol) that sustain the secretory endometrium and provide negative feedback to prevent new follicle recruitment.
Progesterone from the CL peaks around day 21-22 of a 28-day cycle (approximately 7 days after ovulation), reaching 10-25 ng/mL in a healthy luteal phase.
Measurement of serum progesterone on day 21 (or 7 days post-ovulation confirmed by LH testing) is the standard method for confirming ovulation.
A progesterone level above 3 ng/mL indicates ovulation occurred; a level above 10 ng/mL suggests adequate CL function.
If pregnancy does not occur:
The CL begins to involute approximately 10-12 days after ovulation.
Without hCG from an implanting embryo, declining LH support triggers structural luteolysis.
Progesterone and estradiol fall.
The low estrogen removes positive progesterone receptor signaling from the endometrium.
PGF2-alpha (prostaglandin F2-alpha) mediates vasoconstriction and cell death in the CL.
The falling hormone levels withdraw endometrial support, and menstruation begins.
If pregnancy occurs:
The embryo's trophoblast cells produce hCG (human chorionic gonadotropin) beginning at implantation (approximately day 6-10 post-ovulation).
hCG, an LH analog, binds the LH/hCG receptor on the CL and rescues it from luteolysis.
The CL continues producing progesterone throughout the first trimester.
By week 8-10, the placenta takes over progesterone production (the luteal-placental shift).
Luteal phase defect (LPD):
A shortened luteal phase (less than 10 days from ovulation to menstruation) or inadequate progesterone production (less than 10 ng/mL at day 21) can impair implantation and early pregnancy.
LPD is associated with hyperprolactinemia, thyroid dysfunction, low LH pulse frequency, and excessive exercise/energy deficit.
Extraovarian Estrogen: Adipose, Brain, And Adrenal
The ovaries are the dominant source of estradiol in premenopausal women, but they are not the only source.
Adipose tissue:
Aromatase (CYP19A1) is highly expressed in adipose tissue and converts circulating androstenedione (primarily from the adrenal) and testosterone to estrone and estradiol.
This peripheral aromatization is the primary source of estrogen in postmenopausal women.
It is also why higher body fat is associated with higher estrogen levels in both pre- and postmenopausal women, and why obesity is a risk factor for estrogen-sensitive cancers (endometrial, breast).
Brain:
Local brain aromatase produces estradiol directly in neurons (neurosteroid estradiol).
Neurosteroid estradiol acts on synaptic plasticity, neuroprotection, and mood regulation.
Adrenal:
The adrenal zona reticularis produces DHEA and DHEA-S, which are converted peripherally to androstenedione and then to estrogen by aromatase.
This adrenal contribution is relatively minor in premenopausal women but becomes proportionally more significant in menopause when ovarian production ceases.
Estrogen Receptors: ERalpha And ERbeta
Estrogens signal through two nuclear receptor isoforms with partially overlapping but often functionally opposing actions:
ERalpha (ESR1):
Dominant mediator of estrogen's effects on the uterus, breast, liver, bone (via osteoblasts), hypothalamus, and pituitary.
ERalpha drives uterine growth, proliferative endometrium, and hepatic SHBG production.
Activates both estrogen response element (ERE)-dependent transcription and ERE-independent signaling (via AP-1, Sp1 sites).
ERE-independent ERalpha signaling is sufficient for the majority of estrogen's negative feedback on the HPG axis.
ERE-dependent ERalpha signaling is required for the positive feedback LH surge and for spontaneous ovulation. R
In the ERalpha/ERbeta heterodimer, ERalpha is the functionally dominant partner.
ERbeta (ESR2):
Expressed most highly in the ovary (granulosa cells), brain, lung, colon, prostate, and bone.
ERbeta often opposes ERalpha: it can suppress ERalpha-driven proliferative responses in the uterus and breast.
In the ovary, ERbeta on granulosa cells regulates follicle maturation and luteinization; ERbeta-deficient mice are subfertile with abnormal folliculogenesis.
In the brain, ERbeta mediates some of estrogen's anxiolytic and neuroprotective effects.
Tissue-specific ER ratios determine estrogen's net effect:
Tissues where ERalpha predominates (uterus, classic female breast) show proliferative responses to estrogen.
Tissues where ERbeta is relatively higher (ovary, lung, colon) show anti-proliferative or modulatory responses.
This receptor ratio is why SERMs (selective estrogen receptor modulators) can simultaneously act as agonists in some tissues and antagonists in others:
Tamoxifen: ERalpha antagonist in breast (anti-cancer), ERalpha partial agonist in uterus (increased endometrial cancer risk), ERalpha agonist in bone (protective against osteoporosis).
Raloxifene: ERalpha antagonist in breast and uterus, ERalpha agonist in bone.
Mechanism of ER action:
Estradiol diffuses into cells (being lipid-soluble) and binds the LBD of ERalpha or ERbeta.
Ligand binding causes release from heat shock proteins, receptor dimerization (homodimers or ERalpha/ERbeta heterodimers), nuclear translocation, and DNA binding at ERE sequences (5'-GGTCAnnnTGACC-3') in gene promoters.
Co-activators (SRC/p160 family, CBP/p300) are recruited at the ERalpha AF-2 surface, driving histone acetylation and gene transcription.
Rapid non-genomic estrogen signaling also occurs through membrane-associated ER (or GPR30/GPER) that activates MAPK, PI3K/Akt, and calcium signaling within seconds to minutes.
Progesterone Receptor Mechanism
Progesterone signals through two isoforms from the same gene, differing by the presence or absence of the A-domain:
PR-A: shorter, acts as a transcriptional repressor of PR-B (and of ERalpha) in some contexts.
PR-B: longer, the primary transcriptional activator; drives secretory endometrial transformation, mammary gland development, and luteal phase maintenance.
PR-A suppresses PR-B activity and can suppress ERalpha activity in the same cell, which partially explains how progesterone limits estrogen-driven proliferation in the uterus.
The ratio of PR-A to PR-B shifts across tissues and cycle phases and modulates the net progesterone response.
Progesterone receptor expression is induced by estrogen (via ERalpha): progesterone cannot act in a tissue unless estrogen has first primed that tissue by inducing PR expression.
This is the molecular basis for the clinical principle that progesterone supplementation requires adequate estrogen as a foundation.
Perimenopause And Menopause: What Actually Changes
Perimenopause is not simply declining estrogen.
It is a period of erratic and unpredictable ovarian function that often begins 4-10 years before the final menstrual period (on average beginning in the mid-40s). R
What happens in perimenopause:
The ovarian reserve (pool of primordial follicles) diminishes with age.
The remaining follicles are less responsive to FSH.
To recruit and mature a follicle, the pituitary must secrete more FSH.
FSH rises, often with wide fluctuations, reflecting the variable quality of remaining follicles.
Estradiol levels can be normal, low, or paradoxically high (when FSH is elevated enough to hyperstimulate a follicle).
Ovulation becomes erratic and less frequent; luteal phase progesterone production becomes inadequate in cycles where an inferior follicle ovulates.
The result: irregular cycles, variable hormone levels, and symptoms from both estrogen excess (breast tenderness, heavy periods when estradiol is high) and estrogen deficiency (hot flashes, poor sleep, vaginal dryness when estradiol drops).
This chaotic phase can last 4-8 years.
Menopause:
Defined as 12 consecutive months of amenorrhea after the final menstrual period, occurring on average at age 51-52 in the US.
After menopause, ovarian follicle activity essentially ceases.
Estradiol drops to approximately 10-20 pg/mL (from 30-400 pg/mL premenopausally).
FSH rises to greater than 40 IU/L (from approximately 3-10 IU/L in the follicular phase).
Estrone becomes the dominant circulating estrogen (produced by adrenal-derived androstenedione aromatization in adipose tissue).
Progesterone falls to negligible levels (less than 0.5 ng/mL).
Testosterone and DHEA also decline with menopause, though more gradually and less dramatically than estradiol.
The adrenal androgen DHEA peaks in the mid-20s and declines continuously (adrenopause), reaching approximately 20-30% of peak levels by age 70, independent of menopause.
Consequences of postmenopausal estrogen deficiency:
- Vasomotor symptoms (hot flashes, night sweats): from hypothalamic thermoregulatory dysregulation driven by NKB/kisspeptin neuron hyperactivity without estrogen modulation
- Genitourinary syndrome of menopause (vaginal atrophy, dryness, dyspareunia, urinary symptoms): from ERalpha-dependent maintenance of vaginal and urethral epithelium
- Bone loss: accelerated by 3-5% per year in the first 5 years postmenopause from loss of estradiol's suppression of osteoclast activity
- Cardiovascular risk increase: from loss of estradiol's vasoprotective effects on endothelium and lipid profiles
- Cognitive changes: neurosteroid estradiol in the brain is lost; risk of Alzheimer's disease increases with earlier menopause onset
PCOS: A Disease Of LH-Driven Theca Cell Excess
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder in women of reproductive age, affecting approximately 10-15%.
Understanding PCOS requires understanding the two-cell hypothesis and what happens when the LH signal to theca cells is chronically elevated.
The core PCOS pathophysiology:
In PCOS, GnRH pulse frequency is elevated (due to reduced progesterone and steroid feedback on GnRH neurons, partly from insulin resistance disrupting the neuroendocrine axis).
Elevated GnRH frequency biases the pituitary toward LH secretion over FSH secretion.
The resulting elevated LH/FSH ratio over-stimulates theca cells.
Theca cells produce excess androstenedione and testosterone.
But granulosa cells, which depend on FSH to upregulate aromatase, are relatively understimulated.
The imbalance: more androgen substrate arriving in granulosa cells than aromatase can convert to estrogen.
Excess androgens prevent follicle maturation, cause follicle arrest, and promote follicular atresia.
Arrested follicles accumulate (the "polycystic" appearance on ultrasound reflects arrested antral follicles, not true cysts).
No dominant follicle emerges; ovulation fails; progesterone is not produced; negative feedback remains inadequate; LH stays elevated. R
Insulin resistance makes it worse:
Hyperinsulinemia directly stimulates theca cell androgen production (insulin amplifies LH signaling in theca cells via IGF-1 receptor cross-talk).
Hyperinsulinemia also suppresses hepatic SHBG production, increasing free androgen levels beyond what total testosterone alone reveals.
This is why metformin (and GLP-1 receptor agonists) improve PCOS: reducing insulin resistance reduces both hyperinsulinemia-driven androgen production and SHBG suppression.
Letrozole and PCOS:
Aromatase inhibitors (letrozole) are now the first-line ovulation induction agent in PCOS, having displaced clomiphene in most guidelines.
By blocking estrogen production, letrozole removes the negative feedback on FSH, allowing FSH to rise and recruit dominant follicles without the anti-estrogenic effect on endometrium that clomiphene causes.
What To Stay Away From
- Interpreting cycle hormone levels without knowing the cycle day: estradiol ranges from approximately 30 pg/mL in early follicular phase to 400 pg/mL at the preovulatory peak; a result of 200 pg/mL means something completely different on day 5 vs. day 12; always record cycle day when interpreting reproductive hormones
- Assuming perimenopause means low estrogen throughout: the first years of perimenopause are often characterized by estrogen excess from hyperstimulated follicles (erratic, high FSH-driven E2 surges), not deficiency; progesterone deficiency from anovulatory cycles is often the earlier and more consistent hormonal change, causing irregular and heavy bleeding before estrogen itself declines substantially
- Using standard estradiol immunoassays to interpret female levels at the postmenopausal low end: standard assays designed for the wider range are unreliable below 30-50 pg/mL; postmenopausal and on-treatment estradiol levels require a sensitive LC-MS/MS estradiol assay
- Treating PCOS with progesterone-only or estrogen-only interventions without addressing the LH/insulin axis: progesterone withdrawal bleeds manage symptoms temporarily; the underlying problem is excess LH stimulation of theca cells amplified by insulin resistance; sustainable improvement requires addressing insulin sensitivity, not just cyclically inducing a bleed
- Combining bioidentical hormone therapy without including progesterone in women with an intact uterus: estrogen therapy without progesterone (unopposed estrogen) drives endometrial proliferation and substantially increases endometrial cancer risk; women with a uterus must receive progesterone (natural micronized progesterone or a synthetic progestin) alongside estrogen; this is not optional
Testing
Estradiol (E2): Use the sensitive LC-MS/MS assay for postmenopausal women or monitoring HRT; standard immunoassay is acceptable for reproductive-age women when levels are in the normal mid-cycle range. Interpret with cycle day noted.
FSH and LH: Day 3 FSH and LH (early follicular phase, days 2-4) are the standard assessment of ovarian reserve (FSH) and LH/FSH ratio (PCOS screening); FSH above 10 IU/L on day 3 suggests diminished ovarian reserve; elevated LH/FSH ratio above 2-3 is consistent with PCOS.
Progesterone: Day 21 (or 7 days post-confirmed ovulation) progesterone confirms ovulation and assesses luteal function; above 10 ng/mL indicates adequate corpus luteum function; above 3 ng/mL indicates ovulation occurred.
AMH (Anti-Mullerian Hormone): Produced by granulosa cells of small antral follicles; reflects total ovarian follicle reserve; relatively stable across the cycle, unlike FSH; lower AMH indicates diminished reserve; elevated AMH (above 6-7 ng/mL) is consistent with PCOS and follicle accumulation.
Total and Free Testosterone: Elevated total and free testosterone in women with PCOS, CAH, or adrenal androgen excess; free testosterone (calculated or by equilibrium dialysis) is important because elevated SHBG in some conditions can normalize total T while free T remains elevated.
DHEA-S: Adrenal-derived androgen; elevated in adrenal androgen excess (adrenal PCOS variant, CAH, adrenal tumors); normal DHEA-S with elevated testosterone suggests ovarian rather than adrenal source.
17-Hydroxyprogesterone (17-OHP): Elevated in 21-hydroxylase deficiency (CAH); should be measured early morning (best when drawn before 9 AM, as it peaks with cortisol rhythm); borderline levels require ACTH stimulation test; late-onset CAH is a common mimic of PCOS.
Thyroid panel (TSH, free T4): Hypothyroidism raises SHBG, disrupts LH/FSH secretion, causes anovulation, and contributes to luteal phase defect; should be included in any menstrual irregularity evaluation.
Prolactin: Hyperprolactinemia suppresses GnRH and LH/FSH, causing anovulation and amenorrhea; the most common cause of amenorrhea beyond normal pregnancy that responds to treatment; evaluate in any secondary amenorrhea.
SHBG: Elevated in hyperthyroidism and estrogen excess; reduced in insulin resistance, obesity, androgens, and hypothyroidism; low SHBG increases free androgen availability; important for interpreting total testosterone.
Mechanisms Of Action
Simple:
- The ovary cannot make estrogen with just one cell type; theca cells get LH's signal and make androgens (testosterone and androstenedione), but they have no aromatase to finish the job; granulosa cells get FSH's signal and use their aromatase to convert those androgens into estradiol; take either cell type away, or remove either gonadotropin, and estrogen production fails.
- During the first half of the cycle, rising estradiol tells the brain to make less FSH and LH (negative feedback); but when estradiol stays high enough for long enough, the brain and pituitary suddenly flip their response and estradiol becomes a stimulator, triggering the massive LH surge that causes ovulation. This switch from negative to positive feedback is one of the strangest and most important events in reproductive endocrinology.
- The corpus luteum is a temporary gland built from the ruptured follicle; it produces progesterone for about 14 days and then self-destructs if pregnancy does not occur; its entire existence depends on LH stimulation from the pituitary, but during pregnancy the embryo rescues it by secreting its own LH-like signal called hCG, which is what pregnancy tests detect.
- ERalpha drives proliferative effects of estrogen in the uterus and breast; ERbeta often opposes those effects; the balance between the two receptors in a given tissue determines whether estrogen is pro-proliferative or anti-proliferative there, which is why tamoxifen can be anti-estrogenic in the breast while being partially pro-estrogenic in the uterus and bone.
- PCOS is essentially a runaway LH problem: elevated LH tells theca cells to make too much testosterone, while relatively low FSH means granulosa cells do not have enough aromatase to convert all that testosterone into estrogen; excess testosterone blocks follicle maturation, follicles arrest, ovulation fails, and the whole cycle stalls.
Advanced:
- The estradiol positive feedback switch and kisspeptin/GnRH neuron biology: The switch from negative to positive estradiol feedback requires two distinct neuronal populations and changes in ERalpha signaling mode. KNDy neurons in the arcuate nucleus, which co-release kisspeptin (stimulatory), neurokinin B (stimulatory), and dynorphin (inhibitory), drive GnRH pulse generation. Low-dose estradiol (early follicular phase) increases dynorphin from KNDy neurons, slowing GnRH pulse frequency, biasing pituitary toward FSH. High sustained estradiol (preovulatory) activates a separate population of kisspeptin neurons in the anteroventral periventricular (AVPV) nucleus, which fire in a burst to drive the preovulatory GnRH/LH surge. ERE-dependent ERalpha signaling in AVPV neurons is required for the positive feedback surge; ERE-independent ERalpha signaling mediates much of the negative feedback; these two signaling modes can be genetically dissociated, allowing the demonstration that positive and negative feedback are mechanistically separable events. R
- The two-cell system enzymatic logic and PCOS molecular mechanism: The separation of androgen production (theca) from aromatization (granulosa) creates a substrate-enzyme relay across the follicle basement membrane. In normal folliculogenesis, the FSH receptor on granulosa cells is itself induced by FSH/cAMP signaling, creating a self-amplifying loop as the follicle grows. FOXL2, a forkhead transcription factor expressed in granulosa cells, is an essential driver of CYP19A1 transcription; FOXL2 mutations cause premature ovarian insufficiency. In PCOS, theca cell CYP17A1 activity is intrinsically elevated (independent of LH hyperactivation) due to dysregulation of CYP17A1 gene transcription; theca cells from PCOS patients produce more androgen per unit of LH stimulation than controls. Hyperinsulinemia in PCOS acts synergistically with LH on the IGF-1 receptor on theca cells, amplifying CYP17A1 and HSD3B2 expression and output. This intracellular programming of theca cells toward androgen excess persists in cell culture removed from the body, confirming it is intrinsic to PCOS theca cells rather than simply a response to elevated LH. R
- Allopregnanolone and the GABA-A axis in PMDD and postpartum depression: Progesterone is sequentially reduced by 5alpha-reductase (predominantly SRD5A1 in the brain) to 5alpha-dihydroprogesterone, then by 3alpha-hydroxysteroid dehydrogenase (AKR1C) to allopregnanolone (ALLO). ALLO is a potent endogenous positive allosteric modulator (PAM) of GABA-A receptors, binding at the neurosteroid site (distinct from the benzodiazepine binding site) and greatly enhancing chloride conductance even in the absence of GABA. In the luteal phase, rising progesterone generates rising ALLO, and GABA-A receptor subunit composition adapts: alpha4 and delta subunits (which make receptors particularly ALLO-sensitive) are downregulated in a compensatory response. In PMDD, this compensatory downregulation is thought to be exaggerated, creating a state of reduced GABAergic inhibition as ALLO rises that paradoxically produces anxiety and irritability rather than anxiolysis. Postpartum, the rapid drop in progesterone/ALLO after delivery unmasks upregulated ALLO-sensitive GABA-A subunits that were maintained during pregnancy, producing excessive GABAergic inhibition withdrawal. Brexanolone (synthetic IV ALLO) and zuranolone (oral ALLO analog) treat postpartum depression by directly providing GABA-A PAM activity that the brain's own ALLO can no longer supply during this withdrawal state.
More Research
- The two-cell two-gonadotropin system means that FSH is not interchangeable with LH for ovarian hormone production: FSH drives aromatization in granulosa cells and estrogen synthesis; LH drives androgen substrate production in theca cells; the ratio of LH to FSH determines the androgen-to-estrogen balance in the follicle, which is the central pathophysiological insight in PCOS where excess LH relative to FSH drives androgen excess. R
- Perimenopause begins with progesterone deficiency, not estrogen deficiency: the earliest hormonal change is anovulation from declining follicle quality, which eliminates the corpus luteum's progesterone production while leaving estrogen relatively intact; the erratic estrogen fluctuations and estrogen excess episodes of early perimenopause are driven by FSH-hyperstimulated follicles; understanding this temporal sequence changes the clinical approach to perimenopausal symptoms.
- Postmenopausal estrogen production from adipose aromatase means that postmenopausal estrogen levels are not zero: they range from approximately 10-30 pg/mL from adrenal androstenedione aromatization; this residual estrogen is protective in some contexts (bone, cardiovascular) but pathological in others (obesity-associated breast cancer and endometrial cancer risk, where adipose aromatase drives tissue estrogen excess despite low serum levels).
- AMH is currently the best single biomarker of ovarian reserve: it is produced by small antral follicles, is relatively stable across the menstrual cycle, and correlates directly with the remaining primordial follicle pool; AMH declines years before cycle irregularity begins; it is elevated in PCOS (reflecting follicle accumulation) and suppressed in diminished ovarian reserve; it should be part of any fertility, menstrual irregularity, or perimenopause evaluation. R
- For comprehensive female hormone testing, the minimum informative panel at different cycle phases includes: days 2-4 for FSH, LH, estradiol, AMH (baseline ovarian assessment); day 21 for progesterone (ovulation confirmation and luteal phase adequacy); and SHBG, total testosterone, DHEA-S, 17-OHP, TSH, and prolactin at any cycle day when evaluating for androgen excess, cycle irregularity, or PCOS. Cycle day must always be documented alongside results.
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Methylated B Complex
1 cap/day with food
SAMe
400mg on empty stomach
Resveratrol
250mg/day





