Phase II Detoxification: The Six Conjugation Pathways That Make Toxins Water-Soluble
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.
Phase II detoxification is where the body takes reactive, dangerous intermediates created by Phase I and converts them into water-soluble molecules that can be excreted through bile, urine, and stool.
Without Phase II, every Phase I metabolite would accumulate as a free radical, causing more damage than the original toxin.
In this post, we will discuss all six conjugation pathways, what impairs them, how they overlap with conditions like histamine intolerance and CIRS, how to support them with targeted supplementation, the genetics behind individual variation, and the biochemistry that ties it all together.
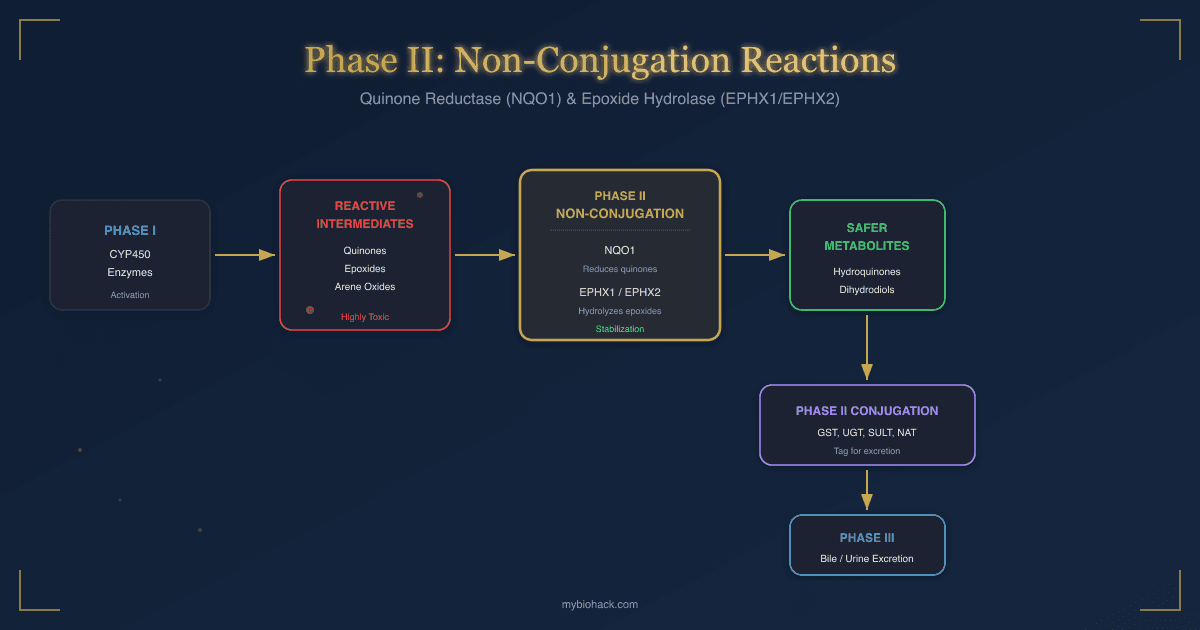
For the non-conjugation Phase II enzymes (quinone reductase and epoxide hydrolase), see the companion post on non-conjugation detox reactions.

Basics Of Conjugation
Conjugation is the process of attaching a polar, water-soluble group to a substrate (usually a Phase I metabolite, drug, hormone, or environmental toxin) so the body can excrete it. R
Phase I creates reactive intermediates through oxidation, reduction, and hydrolysis (mostly via cytochrome P450 enzymes).
These intermediates are often more toxic than the parent compound.
Phase II neutralizes them by covalently attaching a conjugation group (glucuronic acid, glutathione, sulfate, acetyl, methyl, or amino acid) that increases polarity, reduces reactivity, and enables transport into bile or urine. R
There are six major Phase II enzyme families, and each transfers a different molecular group to the substrate.
Key principles of conjugation:
- All six pathways require specific cofactors (nutrient deficiencies directly impair detoxification)
- Competition exists between pathways when a single substrate can be conjugated by more than one enzyme
- Conjugation does not always produce a less toxic product (some sulfation and acetylation reactions generate reactive metabolites)
- Conjugated products must still be transported out of the cell via Phase III transporters (MRP, P-gp, BCRP) for excretion R
- Bile is the primary excretion route for large, glucuronidated, and glutathione-conjugated metabolites
- Urine handles smaller, sulfated and acetylated conjugates
The balance between Phase I activation and Phase II conjugation is critical.
When Phase I runs faster than Phase II (from enzyme induction, genetic polymorphisms, or cofactor depletion), reactive intermediates accumulate and cause oxidative damage to DNA, lipids, and proteins. R
This is the core problem in many chronic illness presentations.
The Six Conjugation Pathways

1. Glucuronidation (UGT Enzymes)
Glucuronidation is the most common Phase II conjugation pathway, accounting for roughly 35% of all drug conjugation reactions. R
The enzyme family responsible is the uridine-diphosphate-glucuronosyltransferases (UGTs), which transfer glucuronic acid from the cofactor UDP-glucuronic acid (UDPGA) to the substrate. R
This makes the substrate significantly more polar and water-soluble, enabling excretion via bile or urine.
UGTs are located in the endoplasmic reticulum of hepatocytes (liver cells) and are also expressed in the gut, kidneys, brain, and skin. R
Substrates glucuronidated by UGTs include:
- Acetaminophen (Tylenol)
- Bilirubin (heme degradation product)
- Estrogens (estradiol, estrone, estriol)
- Morphine and opioids
- NSAIDs (ibuprofen, naproxen)
- Thyroid hormones (T3, T4)
There are four UGT families: UGT1A, UGT2A, UGT2B, and UGT3A. R
The UGT1A family handles bilirubin and many drugs.
The UGT2B family handles steroid hormones and bile acids.
Enterohepatic recirculation is a critical issue with glucuronidation.
Once a glucuronidated conjugate is excreted into bile and reaches the intestine, certain gut bacteria produce an enzyme called beta-glucuronidase that cleaves the glucuronic acid group, liberating the original compound for reabsorption. R
This is why dysbiosis directly impairs glucuronidation efficiency.
Elevated beta-glucuronidase activity from bacterial overgrowth can recirculate estrogens, toxins, and drugs back into the bloodstream, increasing total body burden. R
Calcium-D-glucarate inhibits beta-glucuronidase and supports proper excretion of glucuronidated conjugates. R

2. Glutathione Conjugation (GST Enzymes)
Glutathione S-transferases (GSTs) catalyze the conjugation of reduced glutathione (GSH) to electrophilic substrates. R
Glutathione is the body's master intracellular antioxidant, a tripeptide of glutamate, cysteine, and glycine.
GSTs bind electrophilic compounds (which are inherently reactive and can damage DNA, proteins, and lipids) and attach glutathione to them, neutralizing their reactivity and making them water-soluble for excretion. R
This pathway is the primary defense against:
- Aflatoxin B1 (a carcinogenic mycotoxin produced by Aspergillus)
- Cadmium
- Epoxides (reactive Phase I intermediates from polycyclic aromatic hydrocarbons)
- Lipid peroxidation products (4-hydroxynonenal, malondialdehyde)
- Mercury (organic and inorganic forms)
- Reactive oxygen species (ROS) and reactive nitrogen species (RNS)
GSTs exist in three locations: cytosolic (the largest class, with alpha, mu, pi, theta, sigma, omega, and zeta subfamilies), mitochondrial (kappa class), and microsomal (MAPEG family). R
After glutathione conjugation, the conjugate is further processed through the mercapturic acid pathway: gamma-glutamyl transpeptidase removes glutamate, then a dipeptidase removes glycine, leaving a cysteine conjugate that is N-acetylated to form a mercapturic acid (N-acetylcysteine conjugate). R
This is why NAC (N-acetylcysteine) is so relevant to detoxification: it serves as a precursor to both glutathione synthesis and as the end-product template for mercapturic acid excretion. R
GST activity is inducible by NRF2 activation.
When NRF2 translocates to the nucleus, it binds antioxidant response elements (AREs) in the promoter regions of GST genes, upregulating their expression. R
This is one reason why sulforaphane from cruciferous vegetables is a powerful detoxification inducer.
3. Sulfation (SULT Enzymes)
Sulfotransferases (SULTs) transfer a sulfonate group (SO3−) from the cofactor 3'-phosphoadenosine-5'-phosphosulfate (PAPS) to the hydroxyl or amine group of the substrate. R
Sulfation is a high-affinity, low-capacity pathway.
This means it handles small amounts of substrate very efficiently, but it saturates quickly under high toxic load because PAPS availability is limited by sulfate supply. R
When sulfation saturates, the overflow is typically shunted to glucuronidation.
Substrates sulfated by SULTs include:
- Acetaminophen (at low doses, sulfation handles most of it; at high doses, it overwhelms and glucuronidation/CYP2E1 take over)
- Bile acids
- Catecholamines (dopamine, norepinephrine, epinephrine)
- Cholesterol
- DHEA and steroid hormones
- Estrogens
- Minoxidil (requires sulfation for activation)
- Thyroid hormones
Sulfation is not always detoxifying.
Certain substrates become more reactive after sulfation, a process called bioactivation. R
For example, sulfation of hydroxymethyl polycyclic aromatic hydrocarbons generates highly reactive sulfo-conjugates that bind DNA and initiate carcinogenesis. R
N-hydroxy aromatic amines (from cooked meat heterocyclic amines) are also bioactivated by SULT1A1 to form DNA-reactive intermediates. R
Sulfation depends on adequate dietary sulfur and the amino acids cysteine and methionine (as precursors to sulfate and PAPS). R
Sulfation also plays a role in maintaining the glycocalyx, the sulfated proteoglycan layer lining blood vessels.
Impaired sulfation contributes to glycocalyx degradation and endothelial dysfunction.
4. Acetylation (NAT Enzymes)
N-acetyltransferases (NATs) transfer an acetyl group from acetyl-CoA to the amine group (NH2) of aromatic amines and hydrazines. R
Unlike most Phase II reactions, acetylation does not increase water solubility.
It works by neutralizing the reactive amine group, blocking its ability to form DNA adducts and preventing carcinogenesis. R
The acetylated product is still excreted, but through mechanisms that depend on the balance between acetylation and oxidation rather than polarity alone.
There are two NAT isoforms in humans: NAT1 and NAT2. R
NAT2 is the more clinically relevant because it metabolizes the majority of amine drugs (isoniazid, hydralazine, procainamide, sulfonamides, caffeine). R
Substrates acetylated by NATs include:
- Aromatic amine carcinogens (beta-naphthylamine, 4-aminobiphenyl, benzidine)
- Caffeine (partially, via NAT2)
- Dapsone
- Hydralazine
- Isoniazid (tuberculosis drug)
- Para-aminosalicylic acid (PABA)
- Procainamide
- Sulfonamide antibiotics
Acetylation can also produce bioactivation.
When CYP1A2 (Phase I) oxidizes an aromatic amine to its N-hydroxy derivative, and then O-acetyltransferase activity (catalyzed by the same NAT enzymes) transfers an acetyl group to the oxygen, it creates a highly reactive N-acetoxyarylamine that binds DNA. R
This is one mechanism linking slow acetylator status to increased bladder and colorectal cancer risk. R
The cofactor acetyl-CoA is produced by mitochondria from pyruvate metabolism, fatty acid oxidation, and amino acid catabolism.
Mitochondrial dysfunction therefore directly impairs acetylation capacity.
Pantothenic acid (vitamin B5) is required for CoA synthesis and is a critical cofactor for this pathway. R
5. Methylation (Methyltransferases)
Methyltransferases transfer a methyl group (CH3) from the universal methyl donor S-adenosylmethionine (SAM-e) to oxygen (O-methylation), nitrogen (N-methylation), or sulfur (S-methylation) atoms on the substrate. R
Like acetylation, methylation does not typically increase water solubility.
Instead, it alters biological activity, often inactivating signaling molecules and facilitating their clearance through downstream pathways.
The major methyltransferases are:
- COMT (catechol-O-methyltransferase): inactivates catecholamines (dopamine, norepinephrine, epinephrine), catechol estrogens, and catechol drugs (L-DOPA) R
- HNMT (histamine N-methyltransferase): the primary intracellular histamine degradation enzyme in the brain and peripheral tissues R
- INMT (indolethylamine N-methyltransferase): methylates tryptamine, serotonin, and other indoleamines R
- NNMT (nicotinamide N-methyltransferase): methylates nicotinamide (vitamin B3) and other pyridines R
- TMT (thiol S-methyltransferase): S-methylates thiol drugs (captopril, D-penicillamine) and thiol metabolites R
- TPMT (thiopurine S-methyltransferase): methylates thiopurine drugs (azathioprine, 6-mercaptopurine) used in autoimmune disease and cancer R
Methylation capacity depends on the methylation cycle: methionine to SAM-e to SAH to homocysteine, recycled back to methionine by methionine synthase (B12-dependent) or BHMT (betaine-dependent). R
Every methylation reaction consumes one SAM-e molecule and produces S-adenosylhomocysteine (SAH), which is a potent inhibitor of most methyltransferases. R
When the methylation cycle is impaired (MTHFR variants, B12/folate deficiency, BH4 depletion), SAH accumulates and inhibits COMT, HNMT, and other methyltransferases simultaneously.
This is one mechanism linking methylation dysfunction to elevated catecholamines, histamine intolerance, and impaired xenobiotic clearance.
Specific examples of methylation in detoxification:
- Arsenic detoxification requires sequential methylation: arsenite to monomethylarsonic acid to dimethylarsinic acid (the excretable form), catalyzed by arsenic methyltransferase (AS3MT) using SAM-e R
- Mercury undergoes methylation by gut bacteria, converting inorganic mercury to more toxic methylmercury (this is detrimental methylation, not detoxification) R
- COMT methylation of 4-hydroxy catechol estrogens prevents them from forming reactive quinones that generate ROS and DNA damage R
6. Amino Acid Conjugation (Amino Acid Conjugases)
Amino acid conjugation attaches an amino acid (primarily glycine, but also glutamine, taurine, arginine, and ornithine) to the acyl-CoA thioester of a carboxylic acid substrate. R
This pathway is unique for two reasons:
- The initial activation step occurs in mitochondria via acyl-CoA ligases that require acetyl-CoA (specifically ATP, CoA, and the carboxylic acid substrate form an acyl-CoA intermediate). R
- The conjugation itself is catalyzed by glycine N-acyltransferase (GLYAT) in the mitochondrial matrix (for glycine) or by ACNAT enzymes for other amino acids. R
Substrates conjugated with amino acids include:
- Benzoic acid (from toluene exposure, food preservatives, and gut bacterial metabolism of polyphenols), conjugated with glycine to form hippuric acid R
- Bile acids (conjugated with glycine or taurine for proper emulsification and excretion) R
- NSAIDs and other acyl group drugs
- Phenylacetic acid (from phenylalanine metabolism), conjugated with glutamine to form phenylacetylglutamine R
- Salicylic acid (aspirin metabolite), conjugated with glycine to form salicyluric acid R
Glycine conjugation is the most common amino acid conjugation.
Glycine demand is high: it is used for glutathione synthesis, creatine synthesis, porphyrin synthesis, collagen production, bile acid conjugation, and Phase II conjugation simultaneously. R
Many chronically ill individuals are functionally glycine-deficient because demand exceeds endogenous synthesis and dietary intake.
Taurine conjugation of bile acids is essential for proper biliary excretion of large, lipophilic toxins that have been glucuronidated or glutathione-conjugated.
When taurine levels are low, bile acid conjugation shifts toward glycine conjugation, which produces less efficient bile salt micelles and impairs toxin excretion.
Mitochondrial dysfunction from any cause (biotoxin illness, heavy metals, oxidative stress) directly impairs amino acid conjugation because the activation step requires mitochondrial ATP and CoA.
What Impairs Conjugation

Conjugation failure is rarely about one enzyme.
It is typically a convergence of nutrient depletion, toxic overload, gut dysfunction, and genetic susceptibility that collapses multiple pathways simultaneously.
Nutrient and cofactor depletion:
- B12 and folate deficiency impairs methylation (reduced SAM-e) R
- Glutathione depletion (from chronic oxidative stress, acetaminophen overuse, mycotoxin exposure) impairs GST conjugation R
- Glycine deficiency impairs both amino acid conjugation and glutathione synthesis R
- Magnesium deficiency impairs UGT activity (magnesium is required for UDPGA synthesis) R
- Molybdenum deficiency impairs sulfite oxidase, causing sulfite accumulation and sulfation pathway dysfunction R
- Sulfate and cysteine depletion impairs sulfation (reduced PAPS) R
- Vitamin B5 deficiency impairs CoA synthesis, affecting acetylation and amino acid conjugation R
Toxic overload and substrate competition:
- Alcohol depletes glutathione, NAD+, and SAM-e simultaneously, impairing GST conjugation, methylation, and driving acetaldehyde accumulation R
- High-dose acetaminophen saturates both sulfation and glucuronidation, forcing CYP2E1 to generate the hepatotoxic metabolite NAPQI, which then depletes glutathione catastrophically R
- Multiple toxin exposures create competition for conjugation enzymes, creating a bottleneck where no single toxin is cleared efficiently
- Oral contraceptives induce certain UGTs while depleting B vitamins, folate, and magnesium, creating mixed effects on conjugation capacity R
Gut dysfunction:
- Dysbiosis elevates beta-glucuronidase, deconjugating glucuronidated toxins and estrogens in the gut R
- Leaky gut increases systemic toxin entry, overwhelming conjugation capacity
- Small intestinal bacterial overgrowth (SIBO) impairs taurine and glycine availability through bacterial deconjugation of bile acids R
Liver and mitochondrial dysfunction:
- Chronic liver inflammation reduces UGT, GST, and SULT expression R
- Biotoxin accumulation damages hepatocyte mitochondria, impairing energy-dependent conjugation (acetylation, amino acid conjugation)
- Non-alcoholic fatty liver disease (NAFLD) downregulates multiple Phase II enzymes R
Medications and environmental factors:
- Glyphosate depletes glycine by substituting for it in protein synthesis, and chelates manganese and other minerals needed for conjugation enzymes R
- NSAIDs compete for glucuronidation and amino acid conjugation R
- Proton pump inhibitors reduce magnesium absorption, indirectly impairing glucuronidation R
- Valproic acid depletes carnitine and CoA, impairing acetylation and amino acid conjugation
How Conjugation Overlaps With Other Conditions
Phase II conjugation dysfunction is a thread running through many chronic conditions.
It is rarely discussed as its own diagnosis, but it underlies much of the symptom burden in the following conditions.
Impaired methylation reduces SAM-e, directly reducing COMT, HNMT, TPMT, and AS3MT activity. This creates elevated catecholamines (anxiety, insomnia, racing heart), elevated histamine (flushing, hives, food reactions), impaired thiopurine drug metabolism, and impaired arsenic detoxification. Methylation also feeds into glutathione synthesis (via the transsulfuration pathway), so methylation dysfunction can collapse GST capacity simultaneously. R
Histamine intolerance and mast cell activation:
HNMT is the primary intracellular histamine degradation enzyme, and it depends entirely on SAM-e for its methyl donor. When Phase II methylation is impaired, histamine accumulates. Mast cell degranulation also floods the system with histamine that must be conjugated (both N-methylated by HNMT intracellularly and oxidized by DAO extracellularly). Sulfation is required for degradation of heparin (released alongside histamine from mast cell granules). In MCAS patients, every mast cell degranulation event places acute demand on methylation, sulfation, and glucuronidation pathways simultaneously. R
Biliary dysfunction and gallbladder sludge:
Glucuronidated and glutathione-conjugated metabolites require biliary excretion. When bile flow is sluggish (from gallbladder sludge, bile acid deficiency, or cholestasis), conjugated toxins back up in the liver. This creates feedback inhibition on Phase II enzymes and increases the toxic burden on Phase III hepatocyte transporters. Taurine conjugation of bile acids is critical for bile fluidity, and taurine depletion impairs both bile flow and amino acid conjugation. R
Mycotoxins are potent Phase II substrates. Aflatoxin B1 requires GST conjugation for detoxification, and GSTM1-null individuals have dramatically reduced capacity to clear it. R Ochratoxin A competes for glucuronidation. Trichothecenes deplete glutathione. Chronic mold exposure simultaneously depletes glutathione, SAM-e, and sulfate reserves while increasing the total conjugation demand. NRF2 suppression in CIRS patients further reduces GST and UGT induction capacity.
Bacterial beta-glucuronidase activity deconjugates glucuronidated estrogens, toxins, and drugs. Certain gut bacteria also deconjugate taurine and glycine from bile acids, reducing available amino acids for Phase II. Sulfate-reducing bacteria in the colon consume sulfate that would otherwise be available for PAPS synthesis and sulfation. R
How To Improve Conjugation

The goal is to ensure every conjugation pathway has its required cofactors, the relevant enzymes are properly induced, and the excretion routes (bile, urine, stool) are functioning.
Support glucuronidation:
- Calcium-D-Glucarate inhibits beta-glucuronidase in the gut, preventing deconjugation and recirculation (500 mg, 1-2x daily) R
- Magnesium is required for UDPGA synthesis (glycinate form, 200-400 mg daily) R
- Cruciferous vegetables provide indole-3-carbinol, which upregulates UGT1A1 R
Support glutathione conjugation:
- NAC (N-Acetyl Cysteine) is the rate-limiting precursor for glutathione synthesis (600-1200 mg daily) R
- Liposomal Glutathione provides direct glutathione repletion (250-500 mg daily) R
- Glycine is one of the three amino acids in glutathione and is often the limiting factor (3-5 g daily) R
- Milk Thistle (Silymarin) upregulates glutathione synthesis and has direct hepatoprotective effects (200-400 mg daily) R
- Sulforaphane activates NRF2, which induces GST, UGT, and NQO1 gene expression (from broccoli sprout extract, 30-60 mg daily) R
Support sulfation:
- Taurine provides sulfur, supports bile acid conjugation, and is a direct conjugation amino acid (1-3 g daily) R
- Molybdenum is required for sulfite oxidase, which converts toxic sulfite to sulfate (150-500 mcg daily) R
- NAC also provides cysteine, a sulfur-containing amino acid and PAPS precursor R
- Epsom salt baths provide transdermal magnesium sulfate (both magnesium and sulfate)
Support methylation:
- Methylated B Complex provides methylfolate, methylcobalamin, riboflavin-5-phosphate, and P5P, all required for the methylation cycle R
- SAM-e provides direct methyl donor supplementation (200-400 mg daily on empty stomach, start low and titrate) R
- Magnesium is a cofactor for methionine synthase and multiple methyltransferases R
Support acetylation and amino acid conjugation:
- Glycine is the primary amino acid used in Phase II amino acid conjugation and is chronically depleted in many patients (3-5 g daily) R
- Taurine supports bile acid conjugation and is a secondary amino acid conjugation substrate R
- Methylated B Complex provides vitamin B5 (pantothenic acid), required for CoA synthesis R
Support excretion (binders and bile flow):
- Activated Charcoal binds conjugated toxins in the gut, preventing reabsorption (500-1000 mg, 2 hours away from food and supplements) R
- Zeolite (clinoptilolite) binds heavy metals and mycotoxins in the GI tract R
- Milk Thistle supports bile flow (choleretic effect) and hepatocyte protection R
- Adequate fiber intake supports stool transit and reduces enterohepatic recirculation time
What To Stay Away From
These compounds either deplete conjugation cofactors, overwhelm the pathways, or directly inhibit Phase II enzymes.
- Acetaminophen in doses above 2g/day (saturates sulfation and glucuronidation, depletes glutathione catastrophically) R
- Alcohol (depletes glutathione, NAD+, SAM-e, and directly damages hepatocytes) R
- Charcoal-grilled meats (heterocyclic amines that undergo bioactivation via sulfation and acetylation, generating DNA-reactive intermediates) R
- Chronic NSAID use (competes for glucuronidation and amino acid conjugation pathways)
- Excess fructose (depletes ATP and drives uric acid production, impairing energy-dependent conjugation) R
- Glyphosate residues on non-organic produce (disrupts glycine metabolism and chelates manganese) R
- High-dose turmeric or quercetin without cycling (both inhibit SULT1A1 and can paradoxically impair sulfation at high sustained doses) R
- Processed foods with benzoate preservatives (benzoic acid consumes glycine through conjugation, depleting the glycine pool for other Phase II reactions) R
- Proton pump inhibitors chronically (reduce magnesium absorption, impair glucuronidation) R
Testing
These tests can help identify which conjugation pathways are impaired and what is contributing to the dysfunction.
- Foundation Zoomer: broad-spectrum baseline markers including liver enzymes, inflammatory markers, and nutrient status that reflect overall Phase II capacity
- Cellular Zoomer: measures mitochondrial function, oxidative stress markers, and glutathione status, all of which directly impact GST conjugation and energy-dependent pathways
- Toxin Zoomer: detects environmental toxins (heavy metals, plasticizers, pesticides) and their metabolites, showing what the body is being asked to conjugate
- Mosaic OAT (Organic Acids Test): hippuric acid (glycine conjugation of benzoic acid), pyroglutamic acid (glutathione depletion marker), methylation markers, and mitochondrial intermediates
- Hepatic Detox Profile: directly measures Phase I and Phase II enzyme activity using challenge substrates (caffeine for CYP1A2/NAT2/xanthine oxidase, aspirin for glycine conjugation, acetaminophen for glucuronidation/sulfation/GST conjugation)
- Hepatic Function Panel: liver enzymes (ALT, AST, GGT, ALP, bilirubin) that reflect hepatocyte health and biliary function
- Nutrient Zoomer: measures B12, folate, B6, magnesium, zinc, selenium, cysteine, glutathione, and other cofactors critical for every conjugation pathway
- Methylation Genetics: MTHFR, COMT, CBS, MTR, MTRR, BHMT, and related SNPs that directly affect methylation conjugation and transsulfuration to glutathione
A combination of the Hepatic Detox Profile (functional Phase I/II capacity), the OAT (downstream organic acid metabolites), and the Nutrient Zoomer (cofactor status) provides the most comprehensive picture of conjugation function.
Elevated bilirubin with otherwise normal liver enzymes is a red flag for UGT1A1 dysfunction (Gilbert syndrome). R
Elevated pyroglutamic acid on an OAT is a red flag for glutathione depletion. R
High hippuric acid on an OAT suggests excessive benzoate exposure or glycine depletion (the body is using all available glycine for benzoate conjugation). R
Mechanisms Of Action
Simple
Phase II detoxification works by attaching a chemical group to a toxin, drug, or hormone so the body can dissolve it in water and flush it out.
Phase I makes the toxin reactive (adds a handle).
Phase II sticks a water-soluble tag onto that handle.
Phase III pumps the tagged molecule into bile (for stool excretion) or blood (for kidney/urine excretion).
If Phase II is too slow, the reactive Phase I intermediates accumulate and cause oxidative damage, DNA mutations, and cellular inflammation.
Six different tags exist because different toxins have different chemical structures. Some need glucuronic acid (large, bulky tag), some need glutathione (for reactive electrophiles), some need a sulfate group (small, efficient for hormones and neurotransmitters), some need an acetyl group (for amines), some need a methyl group (for catecholamines and histamine), and some need an amino acid like glycine (for organic acids).
Each tag requires a specific cofactor (vitamin, mineral, or amino acid) to be available. When any of these are depleted, that pathway slows down and toxins back up.
Advanced
Phase II conjugation reactions are catalyzed by superfamilies of transferase enzymes that use activated cosubstrates to form covalent bonds with electrophilic or nucleophilic centers on substrates.
Glucuronidation:
UGTs catalyze an SN2 nucleophilic substitution where the substrate's nucleophilic functional group (OH, NH2, SH, COOH) attacks the C1 carbon of glucuronic acid in UDPGA, displacing UDP. R
The reaction inverts the configuration at C1 from alpha (in UDPGA) to beta (in the glucuronide product).
UGTs are integral membrane proteins of the endoplasmic reticulum, with the active site facing the ER lumen (latency concept: full activity requires membrane disruption or detergent in vitro). R
UDPGA is synthesized in the cytosol from UDP-glucose by UDP-glucose dehydrogenase (requires NAD+), then transported into the ER lumen by the UGT-associated transporter.
Product glucuronides are exported from hepatocytes by MRP2 (canalicular, into bile) and MRP3/MRP4 (basolateral, into blood for renal excretion). R
Glutathione conjugation:
GSTs catalyze the conjugation of the thiol group of reduced glutathione (gamma-Glu-Cys-Gly) to an electrophilic carbon, nitrogen, or sulfur atom on the substrate. R
The key catalytic mechanism involves lowering the pKa of the glutathione thiol from 9.0 to approximately 6.5, generating the thiolate anion (GS−) at physiological pH, which is a potent nucleophile. R
Cytosolic GSTs function as homodimers or heterodimers with a conserved N-terminal domain containing the GSH binding site (G-site) and a variable C-terminal domain containing the hydrophobic substrate binding site (H-site). R
The glutathione conjugate is then processed through the mercapturic acid pathway: gamma-glutamyl transpeptidase (GGT) on the external surface of renal and biliary epithelia cleaves the gamma-glutamyl bond, aminopeptidase M removes glycine, and cytosolic N-acetyltransferase acetylates the remaining cysteine to form the mercapturic acid. R
Export is via MRP1 (basolateral) and MRP2 (canalicular) transporters.
Sulfation:
SULTs catalyze the transfer of the sulfonate group from PAPS to the hydroxyl group of the substrate via a sequential ordered mechanism where PAPS binds first. R
PAPS is synthesized in two steps: ATP sulfurylase condenses ATP and inorganic sulfate to form adenosine-5'-phosphosulfate (APS), then APS kinase phosphorylates APS to form PAPS (at the cost of a second ATP). R
PAPS synthesis is rate-limiting for sulfation capacity and depends on adequate intracellular sulfate (derived from cysteine catabolism, dietary sulfate, and sulfite oxidase activity). R
The product PAP (3'-phosphoadenosine-5'-phosphate) is a competitive inhibitor of SULTs that must be degraded by PAP phosphatase to prevent feedback inhibition.
Acetylation:
NATs catalyze a ping-pong bi-bi mechanism: acetyl-CoA first acetylates a cysteine residue (Cys68) in the NAT active site, releasing CoA, and then the acetyl group is transferred from Cys68 to the substrate amine. R
The catalytic triad is Cys68-His107-Asp122, analogous to cysteine proteases. R
NAT2 polymorphisms alter catalytic activity primarily through effects on protein stability and degradation rates rather than changes to the active site itself (slow acetylator alleles produce unstable protein that is rapidly ubiquitinated and degraded). R
Methylation:
SAM-dependent methyltransferases catalyze the SN2 transfer of the methyl group from the sulfonium center of SAM-e to an electron-rich acceptor atom (O, N, or S) on the substrate, producing the methylated product and SAH. R
COMT uses Mg2+ to coordinate the catechol hydroxyl groups and position the substrate for selective methylation at the meta or para position. R
The SAH product ratio to SAM (the methylation index) determines the thermodynamic driving force for methylation reactions. A low SAM/SAH ratio (below approximately 4:1) indicates methylation insufficiency and correlates with reduced methyltransferase activity across the board. R
Amino acid conjugation:
The two-step reaction involves: (1) mitochondrial medium-chain acyl-CoA ligase (HXM-A and HXM-B) activating the carboxylic acid substrate with CoA in an ATP-dependent reaction to form an acyl-CoA thioester, and (2) GLYAT (glycine N-acyltransferase) transferring the acyl group from CoA to the alpha-amino group of glycine via a ping-pong mechanism. R
GLYAT uses a catalytic His and Asp residue pair to deprotonate the glycine alpha-amino group, making it nucleophilic for attack on the acyl-CoA thioester bond.
The mitochondrial localization of both activation and conjugation steps means that any insult to mitochondrial membrane integrity or matrix function (from biotoxins, heavy metals, or oxidative stress) will directly reduce amino acid conjugation capacity.
Genetics

Genetic variation in Phase II enzymes creates enormous individual differences in conjugation capacity, drug responses, and susceptibility to environmental toxins and cancer.
UGT1A1 and Gilbert Syndrome
UGT1A1 is the sole enzyme that glucuronidates bilirubin.
The most common variant is UGT1A1*28 (rs8175347), a promoter polymorphism where the normal (TA)6 repeat in the TATA box is expanded to (TA)7. R
This reduces UGT1A1 transcription by approximately 70%.
Homozygous UGT1A1*28 causes Gilbert syndrome, a benign condition affecting 5-10% of the population characterized by intermittent unconjugated hyperbilirubinemia (elevated indirect bilirubin) triggered by fasting, stress, illness, or menstruation. R
While Gilbert syndrome itself is considered benign, reduced UGT1A1 activity also impairs glucuronidation of drugs (irinotecan toxicity is dramatically increased in UGT1A1*28 homozygotes) and estrogens. R
Mildly elevated bilirubin actually has antioxidant benefits (bilirubin is a potent free radical scavenger), and Gilbert syndrome has been associated with reduced cardiovascular disease risk. R
Other UGT polymorphisms (UGT1A6, UGT2B7, UGT2B15) affect metabolism of acetaminophen, NSAIDs, and androgens respectively.
GSTM1 and GSTT1 Null Genotypes
GSTM1 (mu class) and GSTT1 (theta class) are unusual because a large proportion of the population carries homozygous gene deletions (null genotype), meaning they produce zero functional enzyme. R
GSTM1-null frequency is approximately 50% in Caucasians, 50% in Asians, and 25% in Africans. R
GSTT1-null frequency is approximately 20% in Caucasians and 50% in Asians. R
GSTM1-null individuals have dramatically reduced capacity to detoxify aflatoxin B1 epoxide, polycyclic aromatic hydrocarbon diol-epoxides, and certain pesticide intermediates. R
GSTM1-null combined with GSTT1-null is a double deletion that significantly increases risk for lung, bladder, and colorectal cancer, especially in smokers and individuals with high occupational toxin exposure. R
GSTP1 (pi class) has a common variant, Ile105Val (rs1695), that reduces catalytic efficiency for polycyclic aromatic hydrocarbons but increases activity toward certain drug substrates. R
For patients with GST null genotypes, aggressive support of glutathione status (NAC, liposomal glutathione, sulforaphane for NRF2 induction of remaining GST isoforms) is essential.
NAT1 and NAT2 Slow Acetylators
NAT2 has over 100 identified alleles, broadly classified as rapid, intermediate, and slow acetylator phenotypes. R
Approximately 40-70% of Caucasians and Africans are slow acetylators (homozygous for slow alleles), compared to 10-30% of East Asians. R
The most common slow alleles are NAT2*5B (rs1801280, Ile114Thr), NAT2*6A (rs1799930, Arg197Gln), and NAT2*7B (rs1799931, Gly286Glu). R
Clinical implications of slow acetylation:
- Increased risk of isoniazid-induced hepatotoxicity and peripheral neuropathy (tuberculosis treatment) R
- Increased risk of sulfonamide hypersensitivity reactions R
- Increased risk of aromatic amine-related bladder cancer in occupational exposure settings (dye workers, rubber industry) R
- Drug-induced lupus from hydralazine and procainamide occurs almost exclusively in slow acetylators R
- Altered caffeine metabolism (slow acetylators clear caffeine more slowly via the NAT2 pathway) R
NAT1 polymorphisms are less well characterized but the slow allele NAT1*14 has been associated with increased risk of cancers from dietary aromatic amines. R
COMT Val158Met
COMT Val158Met (rs4680) is one of the most studied Phase II genetic variants.
The Met/Met genotype produces a thermolabile enzyme with approximately 3-4 fold reduced activity compared to Val/Val. R
Met/Met individuals have higher synaptic dopamine and norepinephrine (because COMT clears them more slowly), which improves prefrontal cortex function and working memory but increases susceptibility to stress-related anxiety, pain sensitivity, and catechol estrogen accumulation. R
Val/Val individuals clear catecholamines rapidly, which can manifest as lower baseline dopamine (anhedonia, reduced motivation) but greater stress resilience. R
The clinical relevance for detoxification: Met/Met individuals have reduced capacity to methylate and inactivate catechol estrogens (2-OH and 4-OH estradiol). R
4-OH catechol estrogens that are not methylated by COMT can be oxidized to quinones that generate reactive oxygen species and form DNA adducts, a mechanism implicated in estrogen-dependent cancers. R
For COMT Met/Met patients, methylation support (adequate B vitamins, magnesium, and SAM-e if tolerated) helps compensate for the reduced enzyme activity.
SULT Polymorphisms
SULT1A1 has a common variant, Arg213His (rs9282861), that reduces enzyme activity and thermal stability. R
Individuals homozygous for the His allele have reduced sulfation capacity for thyroid hormones, estrogens, catecholamines, and many xenobiotics. R
This variant has been associated with increased breast cancer risk in some populations, possibly through reduced sulfation and inactivation of catechol estrogens and environmental estrogens. R
SULT1A3 (catecholamine sulfotransferase) polymorphisms affect platelet sulfation of dopamine and norepinephrine and may contribute to migraine susceptibility and autonomic dysfunction. R
SULT2A1 polymorphisms affect DHEA sulfation and may influence adrenal hormone metabolism.
For patients with SULT polymorphisms, ensuring adequate sulfate supply through dietary sulfur amino acids (cysteine, methionine, taurine) and molybdenum for sulfite oxidase function is important.
More Research
Review articles:
- Jancova P, Anzenbacher P, Anzenbacherova E. Phase II drug metabolizing enzymes. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2010;154(2):103-116. R
- Xu C, Li CY, Kong AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. 2005;28(3):249-268. R
- Allocati N, Masulli M, Di Ilio C, Federici L. Glutathione transferases: substrates, inhibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis. 2018;7(1):8. R
- Gamage N, Barnett A, Hempel N, et al. Human sulfotransferases and their role in chemical metabolism. Toxicol Sci. 2006;90(1):5-22. R
- Hein DW. N-acetyltransferase SNPs: emerging concepts serve as the paradigm for understanding complexities of personalized medicine. Expert Opin Drug Metab Toxicol. 2009;5(4):353-366. R
- Weinshilboum R. Pharmacogenomics: catechol O-methyltransferase to thiopurine S-methyltransferase. Cell Mol Neurobiol. 2006;26(4-6):539-561. R
- Knights KM, Sykes MJ, Miners JO. Amino acid conjugation: contribution to the metabolism and toxicity of xenobiotic carboxylic acids. Expert Opin Drug Metab Toxicol. 2007;3(2):159-168. R
Clinical genetics resources:
- PharmGKB (pharmacogenomics knowledge base): pharmgkb.org
- CPIC (Clinical Pharmacogenetics Implementation Consortium) guidelines for UGT1A1, NAT2, TPMT: cpicpgx.org
Conjugation pathway substrates and testing:
- The Hepatic Detox Profile (caffeine/aspirin/acetaminophen challenge) is the gold standard functional test for Phase I/II conjugation capacity in clinical practice.
- Organic acids testing (hippuric acid, pyroglutamic acid, methylation markers) provides indirect evidence of amino acid conjugation and glutathione status.
- Genetic testing for UGT1A1, GSTM1, GSTT1, NAT2, COMT, and SULT1A1 variants helps identify fixed (non-modifiable) conjugation bottlenecks that require lifelong compensatory strategies.
- For the non-conjugation Phase II enzymes (quinone reductase and epoxide hydrolase) that work alongside these conjugation pathways, see Phase II Non-Conjugation Reactions.
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Spore-Based Probiotics
1 cap with food
L-Glutamine
5g 2x/day on empty stomach
Butyrate
300mg 2x/day with meals