Phase II Detoxification: Non-Conjugation Reactions (Quinone Reductase and Epoxide Hydrolase)
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.
Phase II detoxification is typically associated with conjugation reactions like glucuronidation, sulfation, and glutathione conjugation, but there is a parallel set of Phase II enzymes that do not conjugate anything.
These non-conjugation reactions directly modify reactive intermediates produced by Phase I to make them safer, more water-soluble, and easier for conjugation enzymes to process downstream.
The two major non-conjugation Phase II enzyme families are quinone reductase (NQO1) and epoxide hydrolase (EPHX1/EPHX2).
In this post, we will discuss what these enzymes do, how they fit into the detox pipeline, what impairs them, and how to support them.
For the six conjugation pathways (glucuronidation, glutathione conjugation, sulfation, acetylation, methylation, and amino acid conjugation) that work downstream of these enzymes, see the companion post on Phase II conjugation reactions.

Basics Of Non-Conjugation Detoxification
Phase II detoxification includes two broad categories of reactions: conjugation and non-conjugation. R
Conjugation reactions attach a bulky, water-soluble group (glucuronic acid, sulfate, glutathione, acetyl, methyl, or an amino acid) to a Phase I metabolite, making it large enough and polar enough for excretion through bile or urine.
Non-conjugation reactions do not attach anything.
Instead, they chemically reduce or hydrolyze the reactive intermediate directly, converting a dangerous functional group into a less reactive one. R
Quinone reductase performs a two-electron reduction of quinones, bypassing the toxic semiquinone radical intermediate that one-electron reduction would create. R
Epoxide hydrolase adds water across an epoxide ring, breaking it open to form a dihydrodiol, which is stable, less reactive, and primed for conjugation. R
Both of these reactions serve the same purpose: neutralizing unstable Phase I products before they can damage DNA, proteins, or lipid membranes.
Without functional non-conjugation enzymes, reactive intermediates accumulate and overwhelm the conjugation machinery, creating a bottleneck that drives oxidative stress and tissue damage.
How Non-Conjugation Fits Into The Detox Pipeline
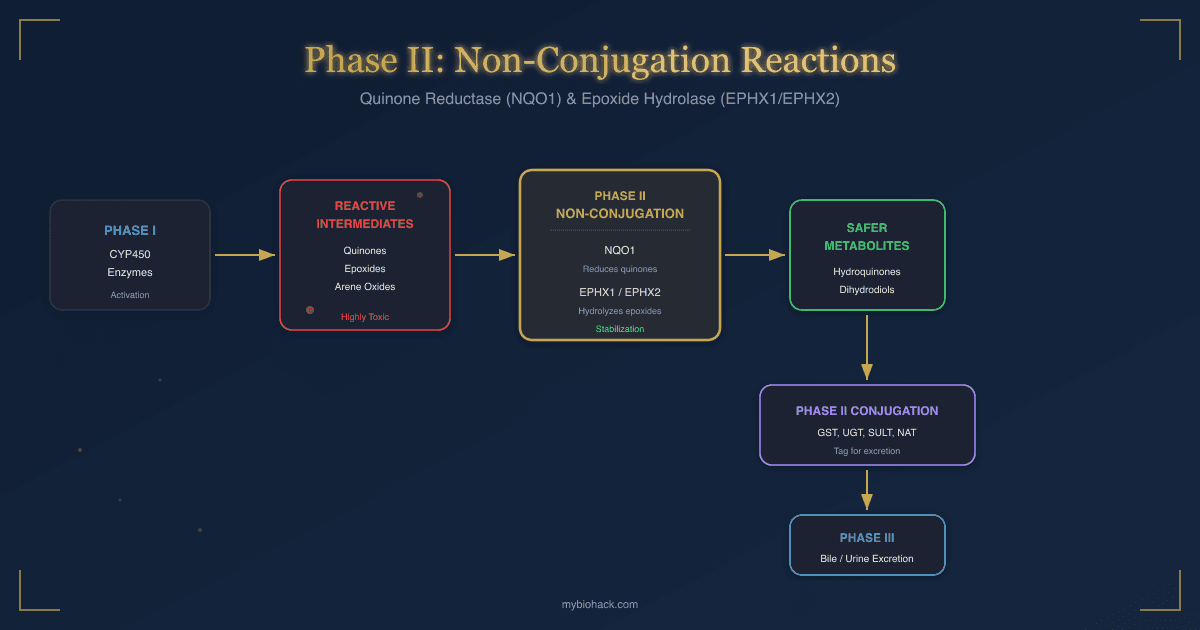
The full detoxification sequence runs through three phases, and non-conjugation reactions occupy a critical middle position.
Phase I: Activation
Phase I CYP450 enzymes add or expose reactive functional groups (hydroxyl, epoxide, quinone) on lipophilic toxins.
This makes the molecule more reactive, not less.
Phase I metabolites are often more dangerous than the parent compound. R
Phase II: Non-Conjugation (Stabilization)
Quinone reductase and epoxide hydrolase intercept the most unstable Phase I products and stabilize them.
NQO1 reduces quinones to hydroquinones, preventing redox cycling.
EPHX1 and EPHX2 hydrolyze epoxides to dihydrodiols, preventing DNA adduct formation.
These stabilized intermediates are now safe enough for the conjugation enzymes to process without damaging surrounding tissue. R
Phase II: Conjugation
Stabilized intermediates are conjugated with glucuronic acid, sulfate, glutathione, acetyl groups, methyl groups, or amino acids.
Conjugation increases molecular weight and water solubility, tagging the metabolite for excretion. R
Phase III: Excretion
Conjugated metabolites are exported from cells by transport proteins (P-glycoprotein, MRP, BCRP) into bile or blood for elimination through feces or urine. R
Bile flow through the gallbladder is a major excretion route for large conjugated metabolites.
The Bottleneck Problem
If Phase I runs faster than non-conjugation and conjugation can keep up, reactive intermediates accumulate.
This is the Phase I/Phase II imbalance that drives toxin-mediated damage in chronic illness.
Non-conjugation enzymes act as a buffer between Phase I output and conjugation input, catching the most reactive intermediates before they cause harm.
When NQO1 or epoxide hydrolase is impaired (by genetics, nutrient deficiency, or toxic load), the conjugation enzymes receive unstabilized, highly reactive substrates that can damage the conjugation machinery itself. R
This creates a cascading failure across the entire detox pipeline.

Quinone Reductase (NQO1)
Quinone reductase (NAD(P)H:quinone oxidoreductase 1, NQO1) is a Phase II enzyme that performs obligate two-electron reduction of quinones to hydroquinones. R
NQO1 is one of the most important NRF2-inducible enzymes in the body and a primary defense against quinone-mediated oxidative damage.
How NQO1 Works
Phase I CYP450 enzymes oxidize aromatic compounds (polycyclic aromatic hydrocarbons, benzene metabolites, certain drugs) into quinones. R
Quinones are extremely reactive because they can undergo one-electron reduction by other reductases (like cytochrome P450 reductase), producing semiquinone radicals.
Semiquinone radicals react with molecular oxygen to regenerate the quinone and produce superoxide, creating a destructive cycle called redox cycling. R
Each turn of this cycle generates more reactive oxygen species (ROS) without consuming the quinone, meaning one quinone molecule can produce unlimited oxidative damage.
NQO1 bypasses the semiquinone intermediate entirely by performing a direct two-electron reduction, converting the quinone straight to a stable hydroquinone. R
The hydroquinone is then easily conjugated by glucuronidation or sulfation and excreted.
NQO1 uses NADPH (or NADH) as the electron donor and FAD (riboflavin/vitamin B2) as a bound cofactor. R
NQO1 Substrates
NQO1 processes a wide range of substrates:
- Benzoquinones (from benzene metabolism, industrial exposure)
- Catechol estrogen quinones (from estrogen metabolism by CYP1A1/CYP1B1)
- CoQ10 (ubiquinone to ubiquinol reduction)
- Menadione (vitamin K3)
- Mitomycin C and other quinone-containing chemotherapy drugs
- Naphthoquinones
- Polycyclic aromatic hydrocarbon (PAH) quinones (from cigarette smoke, grilled food, air pollution)
NQO1 and NRF2
NQO1 gene expression is controlled by the antioxidant response element (ARE) in its promoter region, which is activated by the transcription factor NRF2. R
Under normal conditions, NRF2 is bound to its inhibitor KEAP1 in the cytoplasm and rapidly degraded.
When electrophilic stress or oxidative stress occurs, KEAP1 releases NRF2, which translocates to the nucleus and binds the ARE, upregulating NQO1 along with hundreds of other protective genes. R
NQO1 is considered the canonical NRF2 target gene and is used as a biomarker of NRF2 activation in research. R
Compounds that activate NRF2 (sulforaphane, curcumin, resveratrol) potently upregulate NQO1. R
DJ-1/PARK7 stabilizes NRF2 and prevents its degradation, indirectly supporting NQO1 expression.
When NRF2 signaling is impaired (as seen in CIRS and chronic biotoxin accumulation), NQO1 expression drops, leaving quinone substrates to redox cycle unchecked.
NQO1 and CoQ10
NQO1 is the primary enzyme responsible for reducing ubiquinone (oxidized CoQ10) to ubiquinol (reduced CoQ10) outside the mitochondrial electron transport chain. R
Ubiquinol is the active antioxidant form of CoQ10 that protects cell membranes from lipid peroxidation.
Individuals with NQO1 genetic variants (particularly the C609T/Pro187Ser polymorphism) have reduced ability to maintain ubiquinol levels, which compromises both mitochondrial function and membrane antioxidant defense. R
This is why some people respond poorly to standard CoQ10 (ubiquinone) supplementation and require the pre-reduced ubiquinol form.
NQO1 and Cancer Protection
NQO1 protects against cancer through two mechanisms.
First, by preventing quinone-mediated DNA damage from redox cycling. R
Catechol estrogen quinones (produced when CYP1B1 metabolizes estradiol) can form depurinating DNA adducts that initiate breast and prostate cancer.
NQO1 reduces these quinones to stable hydroquinones before they reach DNA. R
Second, NQO1 stabilizes the tumor suppressor protein p53 by preventing its proteasomal degradation (through a mechanism independent of MDM2). R
Loss of NQO1 function (via the C609T polymorphism) is associated with increased risk of bladder, colon, lung, and breast cancers. R

Epoxide Hydrolase (EPHX1/EPHX2)
Epoxide hydrolase is a Phase II enzyme that hydrolyzes epoxides (three-membered oxygen-containing rings) into trans-dihydrodiols by adding water across the epoxide bond. R
Epoxides are produced when Phase I CYP450 enzymes oxidize compounds containing carbon-carbon double bonds (unsaturated bonds).
These epoxides are electrophilic and can alkylate DNA, proteins, and lipids, making their rapid hydrolysis essential.
Two Forms: Microsomal (mEH) vs Soluble (sEH)
There are two main forms of epoxide hydrolase with different locations, substrates, and biological roles. R
Microsomal epoxide hydrolase (mEH, encoded by EPHX1):
- Located in the endoplasmic reticulum, physically close to CYP450 enzymes
- Directly intercepts CYP450-generated epoxides as they are produced
- Primary substrates: arene oxides and aliphatic epoxides from xenobiotic metabolism
- Critical for detoxifying benzo[a]pyrene epoxides (from cigarette smoke, grilled food), aflatoxin B1 8,9-epoxide (from mycotoxins), styrene oxide (industrial solvent), and drug epoxides (carbamazepine, phenytoin)
- Expressed at highest levels in liver, lung, kidney, and GI tract
Soluble epoxide hydrolase (sEH, encoded by EPHX2):
- Located in the cytosol and peroxisomes
- Primary substrates: endogenous epoxides derived from fatty acid metabolism
- Metabolizes epoxyeicosatrienoic acids (EETs) into dihydroxyeicosatrienoic acids (DHETs)
- Also hydrolyzes leukotoxin (epoxide of linoleic acid) and other fatty acid epoxides
- Expressed in liver, kidney, heart, and vascular endothelium
mEH Mechanism and Xenobiotic Detoxification
mEH uses a two-step catalytic mechanism. R
Step 1: A catalytic aspartate residue (Asp226) attacks the epoxide, forming a covalent ester intermediate and opening the ring.
Step 2: A water molecule (activated by His431) hydrolyzes the ester bond, releasing the trans-dihydrodiol product.
The dihydrodiol product has two hydroxyl groups, making it significantly more water-soluble than the parent epoxide and primed for conjugation by glucuronosyltransferases or sulfotransferases. R
mEH sits physically adjacent to CYP450 enzymes in the ER membrane, forming a metabolic unit where epoxides are hydrolyzed almost immediately after formation. R
This proximity is critical because many epoxides (particularly arene oxides) are so reactive that they would damage surrounding tissue within milliseconds if not caught.
mEH and Aflatoxin/PAH Detoxification
Aflatoxin B1 (AFB1) is a mycotoxin produced by Aspergillus molds that is one of the most potent liver carcinogens known.
CYP3A4 and CYP1A2 convert AFB1 to its 8,9-epoxide, which binds to DNA (forming AFB1-N7-guanine adducts) and causes hepatocellular carcinoma. R
mEH hydrolyzes the AFB1 8,9-epoxide to the dihydrodiol, which is then conjugated with glutathione by GST and excreted. R
Individuals with slow mEH variants (EPHX1 Tyr113His) have reduced capacity to clear aflatoxin epoxides and higher rates of aflatoxin-related liver cancer, particularly in regions with high dietary aflatoxin exposure. R
Benzo[a]pyrene (BaP) from combustion (cigarette smoke, charred food, air pollution) follows a similar path: CYP1A1/CYP1B1 generates BaP-7,8-epoxide, mEH converts it to a dihydrodiol, and the dihydrodiol is conjugated and excreted.
However, the BaP dihydrodiol can be further oxidized by CYP1A1 to the ultimate carcinogen benzo[a]pyrene-7,8-diol-9,10-epoxide (BPDE), which forms DNA adducts. R
This means mEH activity is a double-edged sword for PAHs: it detoxifies the first epoxide but can generate a substrate for a second, more dangerous epoxide.
The balance between mEH activity, CYP1A1 activity, and glutathione conjugation determines whether PAH exposure leads to DNA damage or safe excretion.
sEH and EET Metabolism
The soluble form of epoxide hydrolase (sEH, EPHX2) has a very different biological role from mEH.
sEH primarily metabolizes endogenous epoxyeicosatrienoic acids (EETs), which are produced from arachidonic acid by CYP2C and CYP2J epoxygenase enzymes. R
EETs are potent anti-inflammatory, vasodilatory, cardioprotective, and analgesic lipid mediators. R
sEH converts EETs to dihydroxyeicosatrienoic acids (DHETs), which are biologically much less active. R
This means that high sEH activity depletes anti-inflammatory EETs, promoting:
- Chronic inflammation
- Endothelial dysfunction
- Hypertension
- Increased pain sensitivity
- Vascular inflammation
sEH Inhibition as a Therapeutic Target
Because sEH destroys anti-inflammatory EETs, sEH inhibitors are being developed as treatments for hypertension, inflammatory pain, cardiac fibrosis, and renal disease. R
In animal models, sEH inhibition:
- Lowers blood pressure in angiotensin II-dependent hypertension
- Reduces inflammatory pain (synergistic with NSAIDs and COX-2 inhibitors)
- Protects cardiac tissue after ischemia-reperfusion injury
- Reduces renal inflammation and fibrosis
Natural sEH inhibitors include omega-3 fatty acids (EPA and DHA), which compete for sEH binding and also generate their own anti-inflammatory epoxides (EEQs and EDPs). R
This is one reason omega-3 supplementation has broad anti-inflammatory effects beyond the COX pathway.
sEH and the Arachidonic Acid Balance
Arachidonic acid metabolism occurs through three main enzyme pathways, and sEH intersects with all of them:
- COX pathway produces prostaglandins and thromboxanes (inflammatory mediators targeted by NSAIDs)
- LOX pathway produces leukotrienes (involved in mast cell activation and histamine release)
- CYP epoxygenase pathway produces EETs (anti-inflammatory, resolved by sEH into inactive DHETs)
When COX and LOX pathways are overactive (as in chronic SALI), the CYP epoxygenase/EET pathway serves as a counterbalance.
If sEH activity is also high, this counterbalance is eliminated, leaving inflammation unchecked. R

What Impairs Non-Conjugation Pathways
Multiple factors can reduce NQO1 and epoxide hydrolase activity, creating dangerous gaps in the detox pipeline.
Nutrient Deficiencies
- Riboflavin (vitamin B2) is the bound FAD cofactor for NQO1; deficiency directly reduces NQO1 catalytic activity R
- NADPH is the electron donor for NQO1; NADPH depletion (from glutathione recycling overload, G6PD deficiency, or high oxidative stress) limits NQO1 throughput
- Selenium is required for glutathione peroxidase, which works alongside NQO1 to manage oxidative stress; selenium deficiency increases the burden on NQO1 R
- Zinc supports Phase II enzyme expression and NRF2 signaling R
NRF2 Suppression
Any condition that suppresses NRF2 activation will reduce NQO1 expression:
- Chronic biotoxin exposure (mold, heavy metals)
- CIRS (NRF2 pathway is chronically dysregulated)
- High levels of the NRF2 inhibitor BACH1
- Chronic inflammation (NF-kB and NRF2 compete for the CBP/p300 coactivator) R
- Aging (NRF2 responsiveness declines with age)
Toxic Overload
High-dose toxin exposure can overwhelm both NQO1 and mEH capacity:
- Mycotoxin exposure generates massive epoxide loads that saturate mEH
- Heavy metals (mercury, arsenic, cadmium) inhibit Phase II enzymes directly by binding to catalytic sulfhydryl groups R
- Chronic alcohol consumption depletes NADPH and induces CYP2E1 (increasing quinone production while reducing NQO1 capacity)
Gut Dysfunction
The gut is a major site of epoxide hydrolase expression. R
Dysbiosis and intestinal inflammation reduce epithelial cell function, potentially impairing gut-based Phase II detoxification.
Gut bacteria also produce their own short-chain fatty acids (butyrate, propionate) that activate NRF2 signaling in colonocytes, so dysbiosis can indirectly reduce NQO1 expression. R
Genetic Variants
The NQO1 C609T (Pro187Ser, rs1800566) polymorphism produces a protein with near-zero catalytic activity and accelerated degradation.
EPHX1 Tyr113His (rs1051740) reduces mEH activity by approximately 40%.
These variants are covered in detail in the Genetics section.
How Non-Conjugation Overlaps With Other Conditions
Non-conjugation pathway impairment intersects with many chronic conditions.
CIRS and Biotoxin Illness
NRF2 dysregulation is a hallmark of CIRS, directly reducing NQO1 expression.
Mycotoxins generate both quinone and epoxide intermediates that require NQO1 and mEH for clearance.
Biotoxin accumulation overwhelms the non-conjugation buffer, allowing reactive intermediates to damage downstream conjugation pathways.
Chronic Inflammation
SALI increases oxidative stress, depleting NADPH needed for NQO1.
High sEH activity destroys anti-inflammatory EETs, removing a natural brake on inflammation.
Mast cell activation and histamine excess both increase arachidonic acid release, generating more substrates for the CYP epoxygenase/sEH pathway.
Cancer Risk
NQO1 loss (C609T homozygotes) increases risk of quinone-mediated DNA damage from PAHs, catechol estrogens, and benzene metabolites. R
Slow mEH (EPHX1 Tyr113His) increases risk of aflatoxin and PAH-mediated cancers. R
The combination of fast Phase I CYP1A1 with slow mEH and slow NQO1 creates the highest risk profile for environmental carcinogenesis.
Methylation Impairment
Methylation and non-conjugation pathways share the NADPH pool.
When methylation demand is high (SAM-e production requires methionine cycling), NADPH availability for NQO1 can become limited.
Glutathione synthesis (downstream of methylation via the transsulfuration pathway) also requires NADPH for recycling, creating competition between glutathione recycling and NQO1 function.
Heavy Metal Toxicity
Mercury, arsenic, and cadmium bind to sulfhydryl groups on Phase II enzymes, directly inhibiting their catalytic activity.
Mercury is a particularly potent NRF2 disruptor, reducing NQO1 expression at the transcriptional level. R
Vascular and Endothelial Dysfunction
High sEH activity depletes EETs in vascular endothelium, contributing to endothelial dysfunction, hypertension, and impaired glycocalyx integrity. R
How To Improve Non-Conjugation Pathways
Supporting non-conjugation Phase II detoxification requires NRF2 activation, cofactor repletion, and substrate-specific support.
NRF2 Activators (Upregulate NQO1)
Sulforaphane is the most potent dietary NRF2 activator and the most studied NQO1 inducer. R
Broccoli sprouts contain the highest concentration of the precursor glucoraphanin.
See The 15+ Benefits of Broccoli Sprouts and Sulforaphane for full mechanisms.
Curcumin (phosphatidylcholine-bound for absorption) activates NRF2 via electrophilic modification of KEAP1 cysteine residues. R
Resveratrol activates NRF2 and has direct quinone reductase-inducing activity in cell culture models. R
Quercetin (phytosome form for absorption) activates NRF2 and also stabilizes mast cells, providing dual benefit for individuals with both detox impairment and histamine issues. R
Glutathione Support
NAC (N-acetyl cysteine) provides cysteine for glutathione synthesis and also directly scavenges reactive intermediates that NQO1 and mEH may miss. R
Liposomal Glutathione replenishes the glutathione pool that works alongside NQO1 and mEH in the detox pipeline. R
Milk Thistle (Silymarin) protects liver cells, supports glutathione levels, and has NRF2-activating properties.
See The 18+ Benefits of Milk Thistle for full mechanisms.
CoQ10 Support (For NQO1 Variants)
Ubiquinol (reduced CoQ10) bypasses the NQO1-dependent reduction step entirely, delivering pre-reduced CoQ10 directly. R
This is particularly important for NQO1 C609T carriers who cannot efficiently reduce ubiquinone to ubiquinol.
Standard CoQ10 (ubiquinone) supplements rely on NQO1 for activation, making them less effective in individuals with NQO1 variants.
sEH Modulation (Support EET Levels)
Omega-3 Fish Oil (EPA/DHA) competes with arachidonic acid for sEH binding and generates anti-inflammatory EEQ and EDP epoxides. R
Higher omega-3 intake shifts the sEH substrate pool away from EET degradation.
Cofactor Repletion
Magnesium (glycinate form) is required for hundreds of enzymatic reactions including those in the NADPH regeneration pathway. R
Vitamin C (liposomal for absorption) supports the antioxidant network that works alongside NQO1, recycling other antioxidants and scavenging free radicals that escape non-conjugation enzymes. R
Riboflavin (vitamin B2) is the bound FAD cofactor for NQO1 and should be repleted in anyone with detox impairment.
Protocol Notes
Start with NRF2 activators and cofactors before adding direct glutathione support.
Individuals with CIRS or NRF2 sensitivity may need to start NRF2 activators at very low doses and titrate slowly, as rapid NRF2 activation can transiently increase detox activity and mobilize stored toxins.
What To Stay Away From
These factors impair non-conjugation detox and should be minimized.
- Alcohol depletes NADPH, induces CYP2E1 (increasing quinone production), and directly damages hepatic Phase II enzymes R
- Charred and smoked foods are high in polycyclic aromatic hydrocarbons (PAHs) that generate epoxide and quinone intermediates R
- Cigarette smoke is the single largest source of PAH and benzene exposure, generating massive quinone and epoxide loads R
- Excess omega-6 fatty acids increase arachidonic acid availability, driving more EET production that sEH then degrades, amplifying the inflammatory cycle R
- High-dose isolated antioxidants (synthetic vitamin E, synthetic beta-carotene) can paradoxically suppress NRF2 by removing the oxidative signal needed for NRF2 activation R
- Mold-contaminated environments generate chronic mycotoxin exposure that overwhelms both mEH and NQO1 capacity while simultaneously suppressing NRF2 R
- Pesticide exposure (organophosphates, organochlorines) generates epoxide intermediates that compete with endogenous substrates for mEH R
- Plastic-packaged food heated in plastic releases BPA and phthalates that require Phase II metabolism, adding unnecessary load to the system
Testing
The following lab tests can assess non-conjugation pathway function, cofactor status, and toxic burden.
Cellular Zoomer measures mitochondrial function, oxidative stress markers, and CoQ10 status, which are directly affected by NQO1 activity.
Toxin Zoomer detects environmental toxins (PAHs, benzene metabolites, phthalates) that require NQO1 and mEH for clearance.
Mosaic OAT (Organic Acids Test) evaluates organic acid metabolites that reflect Phase I/II detox capacity, oxidative stress, and mitochondrial function.
Hepatic Detox Profile measures Phase I and Phase II enzyme activity directly, including markers that reflect non-conjugation pathway capacity.
Nutrient Zoomer assesses cofactor status (B2/riboflavin, selenium, zinc, CoQ10, glutathione) required for NQO1 and mEH function.
Hepatic Function Panel evaluates liver function markers that indicate Phase II enzyme health.
Genetic testing for NQO1 C609T and EPHX1 Tyr113His/His139Arg variants is available through most comprehensive genomic panels (23andMe raw data can be interpreted for these SNPs).
Mechanisms Of Action
Simple
NQO1 catches quinones (toxic products of Phase I) and performs a two-step electron transfer that converts them into harmless hydroquinones, skipping the dangerous radical intermediate that would otherwise spin off unlimited free radicals.
Epoxide hydrolase catches epoxides (reactive three-membered rings produced when Phase I enzymes oxidize double bonds) and cracks them open with water, producing stable dihydrodiols that the body can tag and excrete.
Both enzymes sit between Phase I output and Phase II conjugation input, acting as a safety filter that catches the most reactive intermediates before they damage DNA and cell membranes.
When these enzymes are impaired (by genetics, nutrient deficiency, or toxic overload), reactive intermediates bypass the safety filter and either damage the conjugation enzymes directly or escape into the body and cause oxidative tissue damage.
Advanced
NQO1 catalytic mechanism:
NQO1 is a homodimeric flavoprotein (each subunit contains one FAD molecule) that catalyzes a ping-pong bi-bi reaction. R
In the first half-reaction, NADPH reduces the FAD prosthetic group to FADH2.
NADP+ dissociates.
In the second half-reaction, the quinone substrate binds and accepts both electrons from FADH2 simultaneously, producing the hydroquinone product without releasing a semiquinone radical intermediate. R
This obligate two-electron mechanism is what distinguishes NQO1 from other cellular reductases (cytochrome P450 reductase, cytochrome b5 reductase) that perform one-electron reductions and generate semiquinone radicals capable of redox cycling with O2 to produce superoxide (O2-). R
The hydroquinone products are substrates for UDP-glucuronosyltransferases (UGTs) and sulfotransferases (SULTs), linking non-conjugation reduction to conjugation excretion.
NQO1 also stabilizes p53 by binding directly to the p53 protein and preventing 20S proteasomal degradation in a ubiquitin-independent manner. R
Dicoumarol (a competitive inhibitor of NQO1 at the NADPH binding site) blocks this p53 stabilization, confirming that NQO1 catalytic activity is required for the protective effect.
mEH catalytic mechanism:
mEH (EPHX1) uses a two-step catalytic mechanism involving Asp226 (nucleophile) and His431 (general base/charge relay). R
Step 1 (alkylation): Asp226 attacks the less hindered carbon of the epoxide ring, forming a covalent alpha-hydroxy ester intermediate.
Step 2 (hydrolysis): His431 (assisted by Asp352 in a charge-relay system) activates a water molecule, which attacks the ester carbonyl, releasing the trans-dihydrodiol product and regenerating the free enzyme. R
The rate-limiting step varies by substrate: for some epoxides, alkylation is rate-limiting; for others (like styrene oxide), hydrolysis of the ester intermediate is rate-limiting.
mEH is oriented in the ER membrane with its active site facing the cytoplasmic side, directly accessible to CYP450-generated epoxides that partition into the ER membrane. R
sEH catalytic mechanism and EET biology:
sEH (EPHX2) is a homodimeric bifunctional enzyme. R
The C-terminal domain contains the epoxide hydrolase activity (using Asp334 as nucleophile, analogous to mEH).
The N-terminal domain contains a phosphatase activity that dephosphorylates lipid phosphates (including lysophosphatidic acid), though the biological significance of this phosphatase is still being characterized. R
EETs (5,6-EET, 8,9-EET, 11,12-EET, 14,15-EET) are produced by CYP2C8, CYP2C9, and CYP2J2 from arachidonic acid.
14,15-EET is the preferred sEH substrate, followed by 11,12-EET. R
EETs activate BK(Ca) channels in vascular smooth muscle (causing vasodilation), suppress NF-kB nuclear translocation (reducing inflammatory gene expression), inhibit VCAM-1 expression on endothelium (reducing leukocyte adhesion), and activate PPARgamma (anti-inflammatory transcription factor). R
sEH converts these to DHETs, which have minimal BK(Ca) channel activity, no NF-kB suppressive effect, and weak PPARgamma activation.
Genetics
NQO1 C609T (Pro187Ser, rs1800566)
This is the most studied NQO1 polymorphism and one of the most consequential detox gene variants.
The T allele (Ser187) produces a protein that is catalytically inactive and rapidly degraded via the ubiquitin-proteasome pathway. R
CT heterozygotes have approximately 3-fold reduced NQO1 activity compared to CC wild type.
TT homozygotes have near-zero detectable NQO1 protein and activity. R
The TT genotype frequency varies by ethnicity: approximately 4-5% in Caucasians, 5% in African Americans, and 20-22% in East Asian populations. R
Clinical associations of TT homozygosity:
- Elevated risk of benzene-related leukemia (particularly acute myeloid leukemia) R
- Increased bladder cancer risk (especially in smokers) R
- Increased colorectal cancer risk R
- Poor response to quinone-based chemotherapy drugs (mitomycin C, RH1) because NQO1 activates these prodrugs R
- Reduced CoQ10 (ubiquinol) recycling capacity R
- Reduced p53 stabilization R
Clinical takeaway: TT carriers should supplement with ubiquinol (not ubiquinone), aggressively support NRF2 through other pathways, avoid quinone-generating exposures (benzene, PAHs), and ensure riboflavin status is optimal to support whatever residual NQO1 activity exists.
EPHX1 Tyr113His (rs1051740)
This exon 3 polymorphism reduces mEH catalytic activity by approximately 40%. R
The His113 variant has lower Vmax for most substrates due to altered protein folding and reduced stability.
Clinical associations:
- Increased lung cancer risk in smokers (slow PAH detoxification) R
- Increased hepatocellular carcinoma risk in aflatoxin-exposed populations R
- Increased susceptibility to COPD R
- Possible increased susceptibility to xenobiotic-induced liver damage
EPHX1 His139Arg (rs2234922)
This exon 4 polymorphism increases mEH activity by approximately 25%. R
The Arg139 variant stabilizes the protein and increases catalytic efficiency.
When combined with the slow Tyr113His variant, the net mEH activity depends on which allele combination is present:
- His113/His139 (slow exon 3, normal exon 4): approximately 40% reduced activity
- His113/Arg139 (slow exon 3, fast exon 4): approximately normal activity (the fast exon 4 partially compensates)
- Tyr113/Arg139 (normal exon 3, fast exon 4): approximately 25% increased activity
EPHX2 Variants
Several EPHX2 polymorphisms affect sEH activity and cardiovascular risk:
Arg287Gln (rs751141): Reduces sEH activity, resulting in higher circulating EET levels.
This variant is associated with reduced risk of coronary heart disease and ischemic stroke in some populations because it preserves anti-inflammatory EETs. R
Lys55Arg (rs41507953): Increases sEH activity, associated with increased risk of coronary artery calcification and cardiovascular events. R
Clinical takeaway: For EPHX2 variants, the interpretation is inverted compared to most detox genes. Reduced sEH activity (Arg287Gln) is generally protective because it preserves anti-inflammatory EETs, while increased sEH activity (Lys55Arg) is a risk factor because it depletes EETs faster.

More Research
Non-conjugation Phase II reactions remain understudied compared to conjugation pathways, and several areas need further research.
The interaction between NQO1 genotype and environmental quinone exposure across different populations needs larger prospective cohort studies. R
sEH inhibitor drugs are in clinical trials for hypertension, inflammatory pain, and diabetic nephropathy, with promising results in animal models but limited human data so far. R
The role of mEH in gut-based detoxification and its interaction with the microbiome is poorly characterized.
The relationship between NQO1 variants, CoQ10 status, and mitochondrial disease severity needs clinical investigation, particularly in populations with high TT homozygosity rates. R
Whether natural sEH inhibitors (omega-3 fatty acids, specific flavonoids) can produce clinically meaningful EET preservation in humans at achievable dietary doses is not yet established. R
The crosstalk between NRF2/NQO1, methylation, and glutathione recycling under conditions of high toxic load needs systems-level modeling to understand how these pathways compensate for and compete with each other in real clinical scenarios.
For the six conjugation pathways (glucuronidation, glutathione conjugation, sulfation, acetylation, methylation, and amino acid conjugation) that process metabolites downstream of quinone reductase and epoxide hydrolase, see Phase II Conjugation Reactions.
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Quercetin
500mg 2x/day
SPM Active (Pro-resolving Mediators)
2 softgels/day
Curcumin (Liposomal)
500mg 2x/day