H. Pylori: What It Is, What It Destroys, And How To Get Rid Of It
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.
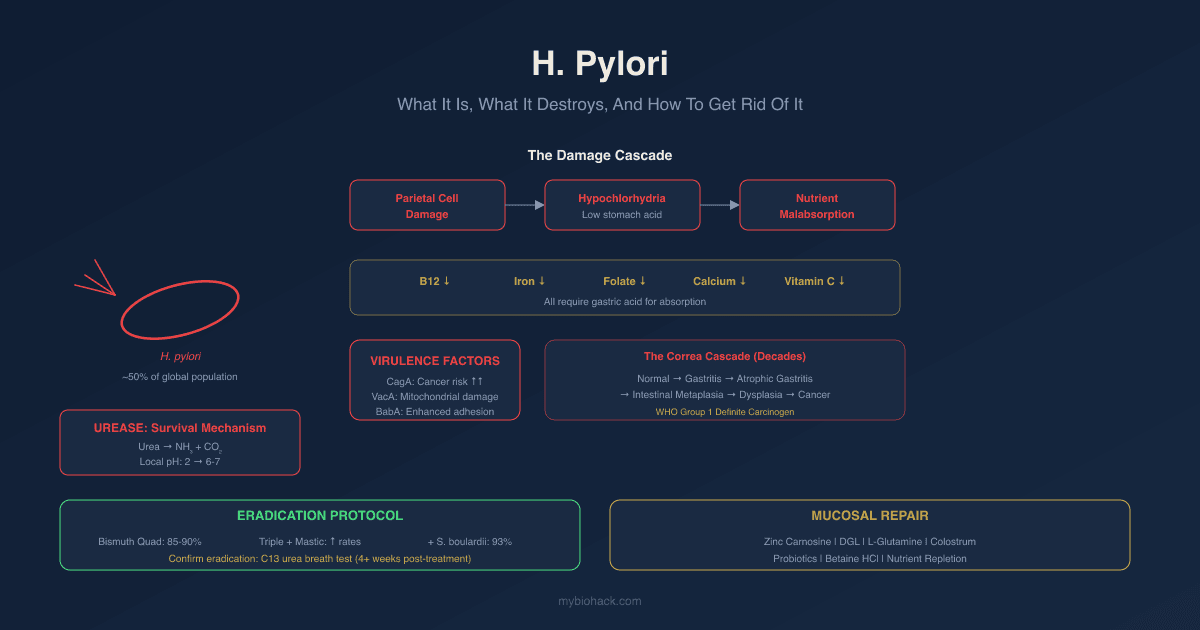
Helicobacter pylori (H. pylori) is the most prevalent chronic bacterial infection in the world, carried by approximately 50% of the global population, and it does far more damage than most people and most doctors appreciate.
In this post, we will discuss what H. pylori is and how it survives stomach acid, what it does to the stomach lining and gastric acid production, the virulence factors that determine how dangerous a particular strain is, the wide range of downstream consequences including nutrient deficiencies, leaky gut, and cardiovascular disease, how to test for it properly, and what the evidence supports for both pharmaceutical and natural eradication protocols.

What Is H. Pylori
H. pylori is a spiral-shaped, gram-negative, microaerophilic bacterium that colonizes the gastric mucosa, the protective lining of the stomach.
It was first identified in 1982 by Australian physicians Barry Marshall and Robin Warren, a discovery that fundamentally changed gastroenterology and earned them the Nobel Prize in 2005.
Before their work, peptic ulcers were believed to be caused by stress, diet, and excess acid.
Marshall famously drank a culture of H. pylori himself to demonstrate the infection, developed gastritis within 3 days, and resolved it with bismuth.
H. pylori infects roughly 50% of the world's population, with higher prevalence in developing countries and in older populations in developed ones. R
Transmission is primarily:
- Fecal-oral (contaminated water or food)
- Oral-oral (sharing utensils, cups, or through direct saliva contact)
- Within family units, where household transmission is common and family members of infected individuals should be considered for testing
Most infections are acquired in childhood and persist indefinitely without treatment.
How H. Pylori Survives And Colonizes
The stomach maintains a pH of approximately 1.5-3.5 under normal conditions.
Nothing should survive there.
H. pylori has evolved a remarkable set of mechanisms to not only survive this environment but to use it as a competitive advantage against other pathogens.
Urease production is the central survival mechanism.
H. pylori produces urease in large quantities.
Urease converts urea (present in gastric secretions and blood) to ammonia and carbon dioxide.
Ammonia is alkaline.
The cloud of ammonia surrounding H. pylori cells locally neutralizes gastric acid, creating a microenvironment with pH close to neutral that allows the bacteria to survive and migrate toward the mucus layer. R
Motility via multiple flagella allows H. pylori to drill through the viscous mucus gel that coats the gastric epithelium. R
Once through, it adheres to gastric epithelial cells via adhesins (including BabA, discussed below) and sets up permanent residence between the mucus layer and the epithelial surface, protected from both acid and immune attack.
The organism preferentially colonizes the gastric antrum (the lower stomach), where the mucus layer is thickest and acid exposure is lower.
From there, in some cases, it migrates to the gastric body, where it can damage the acid-secreting parietal cells directly.
What H. Pylori Does To The Stomach
The core pathological mechanism of H. pylori is progressive destruction of the gastric mucosal architecture, with consequences that cascade far beyond the stomach.
Parietal Cell Damage And Hypochlorhydria
Parietal cells are the specialized gastric epithelial cells that produce hydrochloric acid (via the H+/K+-ATPase pump) and intrinsic factor (required for vitamin B12 absorption).
H. pylori damages parietal cells through both direct cytotoxic mechanisms and indirect immune-mediated inflammation.
The result is hypochlorhydria: reduced gastric acid production.
Severe, prolonged damage progresses to achlorhydria: near-complete absence of gastric acid.
This matters far beyond digestion.
Gastric acid serves as a sterilizing barrier, an activator of pepsin for protein digestion, and the essential medium for converting dietary ferric iron (Fe3+) to the absorbable ferrous form (Fe2+) and for releasing protein-bound vitamin B12 from food. R
When acid is gone, all of these fail simultaneously.
Gastritis And Mucosal Inflammation
H. pylori infection invariably causes gastric mucosal inflammation, ranging from mild non-atrophic gastritis to severe atrophic gastritis.
The inflammatory response involves NF-kB activation, IL-8 and IL-1 release, neutrophil infiltration, and oxidative stress-mediated epithelial damage. R
Inflammation and increased intestinal permeability always occur with H. pylori infection.
H. pylori causes a significant decrease in transepithelial electrical resistance and severe disruption of tight junction proteins ZO-1 and claudin-1 in gastric epithelial cells, establishing the physical basis for leaky gut in infected individuals. R
This drives downstream consequences including food sensitivities, skin issues, and autoimmune conditions.
Eliminating the infection is the root cause intervention. Elimination diets and anti-inflammatory protocols are band-aids when H. pylori is the driver.
The Correa Cascade (Gastric Cancer Progression)
H. pylori is classified as a Group 1 definite carcinogen by the WHO's International Agency for Research on Cancer.
The pathway from infection to gastric cancer follows a well-characterized sequence:
Normal mucosa → Chronic non-atrophic gastritis → Atrophic gastritis → Intestinal metaplasia → Dysplasia → Gastric adenocarcinoma
This progression takes decades and does not occur in all infected individuals.
The probability of progression is modulated by H. pylori strain virulence factors (discussed below), host genetic factors, diet (high salt diet accelerates progression), and whether eradication is achieved before atrophic changes become irreversible. R
Virulence Factors: Not All H. Pylori Is The Same
This is where a positive H. pylori result on a stool test gets stratified into clinical risk categories.
The GI-MAP from Diagnostic Solutions tests for H. pylori quantitatively and reports seven virulence factor genes alongside the main result.
The presence of specific virulence factors determines whether an H. pylori infection is more likely to cause ulcers, gastric cancer, or remain relatively quiescent.
All virulence factor positive results need to be taken seriously and addressed.
BabA (Blood Group Antigen Binding Adhesin)
BabA facilitates the binding of H. pylori to the Lewis b blood group antigen on gastric epithelial cells.
Stronger adhesion means longer contact time, greater virulence factor injection into host cells, more intense inflammation, and greater likelihood of disease progression.
BabA-positive strains are associated with more severe inflammation and increased cancer risk. R
CagA (Cytotoxin-Associated Protein A)
CagA is the most studied and most significant H. pylori virulence factor.
CagA-positive strains carry a pathogenicity island (Cag PAI) encoding a type IV secretion system (T4SS) that injects CagA directly into gastric epithelial cells.
Once inside host cells, CagA is phosphorylated and disrupts multiple signaling pathways, including NF-kB, STAT3, Wnt/beta-catenin, and Hippo, driving uncontrolled cell proliferation and carcinogenesis.
CagA-positive strains significantly increase the risk of gastric cancer and peptic ulcers compared to CagA-negative strains. R
The CagA EPIYA phosphorylation motif pattern (specifically, the number of EPIYA-C repeats in Western strains) determines oncogenic potency: more repeats confer greater CagA activity and higher cancer risk.
VacA (Vacuolating Cytotoxin A)
VacA is a pore-forming toxin secreted by H. pylori that inserts into host cell membranes.
It creates vacuoles (hence the name) in gastric epithelial cells, disrupts mitochondrial membrane potential, induces cytochrome c release, and promotes apoptosis.
VacA-positive strains are associated with gastric cancer, peptic ulcers, and duodenal ulcers through direct mitochondrial damage. R
DupA (Duodenal Ulcer-Promoting Gene A)
DupA is strongly linked to increased risk of duodenal ulcers specifically.
Its mechanism involves promotion of IL-12 production and local inflammation. R
Interestingly, DupA-positive strains appear less likely to cause gastric cancer, suggesting different disease tropism between DupA and CagA.
IceA (Induced by Contact with Epithelium A)
IceA is upregulated when H. pylori contacts gastric epithelial cells.
Meta-analysis has associated IceA with peptic ulcers. R
Its independent contribution to gastric cancer risk is less clear than CagA or VacA.
OipA (Outer Inflammatory Protein A)
OipA increases IL-8 secretion from gastric epithelial cells, driving amplified local inflammation. R
OipA-positive strains are associated with gastric cancer and peptic ulcers and tend to co-occur with CagA positivity.
A Practical Reading Of Virulence Factor Results
The sample GI-MAP result in the course materials showed H. pylori at 3.6e4 (High) with BabA and virD positive but CagA, DupA, IceA, OipA, VacA, and virB all negative.
This represents a moderately virulent strain with adhesion advantage (BabA) but without the high-cancer-risk combination of CagA plus VacA.
BabA positivity alone still warrants full eradication and monitoring.
A result with CagA and VacA both positive warrants urgent eradication, post-eradication surveillance endoscopy, and close monitoring for atrophic changes.
Downstream Consequences: Beyond The Stomach
Nutrient Deficiencies
This cascade is one of the most clinically underrecognized aspects of H. pylori.
Hypochlorhydria from parietal cell damage impairs absorption of: R
- Vitamin B12: B12 from food is bound to dietary proteins. Gastric acid and pepsin cleave B12 from food protein. Once free, B12 binds to intrinsic factor (produced by parietal cells) for absorption in the terminal ileum. H. pylori damages both acid production and parietal cell function, impairing both cleavage steps simultaneously. Prolonged H. pylori infection is a documented cause of vitamin B12 deficiency independent of dietary intake. R
- Iron: Gastric acid is required to convert dietary ferric iron (Fe3+) to ferrous iron (Fe2+), the absorbable form. H. pylori also directly competes with the host for iron, sequestering it for its own growth via lactoferrin receptors and iron-binding proteins, and stimulates hepcidin release which further restricts iron availability. Four meta-analyses have concluded that H. pylori infection is a causative factor in iron deficiency anemia. R
- Calcium and magnesium: Solubility of these polyvalent cations depends on gastric acid. Elevated pH from hypochlorhydria reduces their solubility and absorption. R
- Folate: H. pylori-positive patients show significantly lower serum folate levels than H. pylori-negative patients, partly through increased folate consumption by the bacteria itself and partly through mucosal inflammation reducing folate absorption efficiency. R
- Vitamin C: Gastric juice normally contains concentrated vitamin C, which contributes to H. pylori defense and helps maintain mucosal integrity. H. pylori suppresses gastric vitamin C secretion directly, and the resulting reduction in intragastric vitamin C is one reason the bacteria can colonize so effectively. R
Intestinal Permeability And Systemic Inflammation
H. pylori infection universally increases intestinal permeability.
The inflammatory cascade it generates (IL-8, TNF-alpha, reactive oxygen species) degrades tight junction proteins throughout the gastrointestinal tract. R
This creates the conditions for:
- Increased food sensitivities and allergies (antigens crossing the leaky gut barrier triggering systemic immune sensitization) R
- Skin conditions (eczema, urticaria, rosacea) driven by systemic immune activation and gut-skin axis dysregulation
- Autoimmune conditions, as molecular mimicry between H. pylori antigens and host proteins can trigger autoantibody production
- Dysbiosis, because hypochlorhydria eliminates the primary sterilizing barrier to the upper GI tract, allowing bacterial overgrowth (SIBO) and dysbiotic colonization to proceed unchecked
Thyroid Disease
H. pylori and Hashimoto's thyroiditis have a documented association.
A meta-analysis confirmed that H. pylori infection correlates with both Graves' disease and Hashimoto's thyroiditis, and that eradication of H. pylori reduces thyroid autoantibodies. R
The molecular mimicry mechanism involves CagA-positive strains sharing sequence homology with thyroperoxidase (TPO), explaining why CagA-positive infections carry stronger associations with thyroid autoimmunity. R
H. pylori-induced hypochlorhydria also impairs thyroid hormone absorption, particularly relevant in patients taking oral levothyroxine, where gastric acid aids absorption.
Immune Thrombocytopenic Purpura (ITP)
H. pylori eradication is an evidence-based treatment for H. pylori-associated ITP.
The relationship was first described in 1998, when an Italian group reported significant platelet count increases in 8 of 11 ITP patients following H. pylori eradication. R
The primary mechanism is molecular mimicry: anti-H. pylori CagA antibodies cross-react with platelet surface glycoprotein IIb/IIIa, driving platelet destruction; eradicating the antigen source removes the stimulus for autoantibody production. R
In patients with H. pylori-associated ITP, eradication results in remission or cure of the disease in a substantial proportion of patients, with platelet recovery sustained for 7 or more years in long-term follow-up studies. R
H. Pylori And Cardiovascular Disease
The cardiovascular connections are real, but require honest presentation because the data is conflicting.
Atrial Fibrillation
A significant body of evidence links H. pylori to atrial fibrillation (AF), one of the most common cardiac arrhythmias affecting approximately 2.2 million Americans.
The initial observation came from cardiologists who noticed that a high proportion of AF patients admitted to their department had concurrent gastric complaints and high H. pylori seropositivity. R
In a pilot study, H. pylori seropositivity and CRP were both significantly higher in AF patients compared to controls, with persistent AF patients showing even higher levels than paroxysmal AF patients.
A 943-patient study found that younger patients (under 50) who were H. pylori seropositive had a notably higher relative risk of AF than seronegative peers, though this association weakened after adjusting for confounders including age. R
A 2025 review summarizes proposed mechanisms including systemic inflammation via IL-6 and CRP elevation, gut microbiome disruption affecting the gut-heart axis, metabolic dysregulation (altered LDL and HDL from H. pylori), and a specific molecular mechanism involving autoantibodies against H+/K+-ATPase cross-reacting with cardiac Na+/K+-ATPase, potentially disrupting cardiac ion homeostasis. R
The honest caveat: a meta-analysis found weak correlation between H. pylori and AF that did not reach statistical significance after meta-regression. R
The relationship is associative, biologically plausible, and probably real in a subset of AF patients, but H. pylori is not established as an independent causal factor for AF the way it is for peptic ulcers.
Eradicating H. pylori in a patient with AF and confirmed infection is still appropriate and reasonable.
Coronary Artery Disease
Multiple studies associate H. pylori (particularly CagA-positive strains) with coronary heart disease, atherosclerosis, and adverse lipid profiles (elevated LDL, reduced HDL, elevated triglycerides). R
The proposed mechanism is chronic systemic inflammation driving endothelial damage and accelerating atherosclerotic plaque formation.
Again, the data is associative and conflicting across studies.
The clearest signal is the inflammatory pathway: H. pylori raises CRP and inflammatory cytokines, and chronic systemic inflammation from any cause accelerates cardiovascular disease.
How To Eradicate H. Pylori
Pharmaceutical Triple Or Quadruple Therapy
Standard triple therapy consists of a proton pump inhibitor (PPI) twice daily plus two antibiotics (clarithromycin and amoxicillin, or clarithromycin and metronidazole) for 10-14 days.
Eradication rates with triple therapy have declined to approximately 70-80% in many regions due to rising antibiotic resistance, particularly to clarithromycin and metronidazole. R
Current consensus states that if clarithromycin resistance in a region exceeds 15%, triple therapy should be abandoned in favor of bismuth quadruple therapy or alternative regimens. R
Bismuth quadruple therapy (PPI + bismuth + tetracycline + metronidazole) achieves higher eradication rates (85-90%) and is preferred in areas with high clarithromycin resistance. R
Bismuth has direct anti-H. pylori activity via disruption of cell walls and enzyme inhibition, and also provides mucosal protection.
Confirmation of eradication is essential and should be done at least 4 weeks after completing treatment using a C13 urea breath test or stool antigen test (not serology, which can remain positive for years after successful eradication).
Natural And Functional Medicine Protocol
Natural protocols are appropriate as standalone approaches for lower-burden infections without aggressive virulence factors, as adjuncts to pharmaceutical therapy to increase eradication rates, or for patients who have failed or cannot tolerate antibiotics.
The evidence is honest here: natural agents used alone have significantly lower eradication rates than pharmaceutical triple therapy (typically 30-40% vs 70-90%) in published trials.
They are most effective when combined with pharmaceutical therapy or when used in structured, adequate-duration protocols.
Mastic Gum
Mastic gum is a resin from Pistacia lentiscus, native to the island of Chios in Greece, with centuries of use for stomach complaints.
It has documented direct bactericidal activity against H. pylori at concentrations achievable in the stomach, including activity against clarithromycin-resistant and metronidazole-resistant strains. R
In a randomized pilot study, mastic gum alone (1g/day and 3g/day for 14 days) achieved eradication in approximately 30-38% of patients, compared to 77% for pharmaceutical triple therapy. R
A 2023 study in the American Journal of Clinical Pathology found that adding mastic gum to modified triple therapy significantly improved eradication rates compared to triple therapy alone. R
Mastic gum's value is greatest as an adjunct to pharmaceutical protocols, or in structured extended natural protocols run for 60-90 days rather than the 14-day window used in most trials.
Dose: 1-2g daily between meals for at least 60 days in a natural protocol.
DGL (Deglycyrrhizinated Licorice)
DGL is licorice root with the glycyrrhizin removed (to eliminate the blood pressure-raising, cortisol-sparing effects of glycyrrhizin).
DGL inhibits H. pylori growth and has direct anti-adhesion effects (preventing H. pylori from binding gastric mucosa via BabA adhesins).
A randomized clinical trial demonstrated that adding licorice to a standard clarithromycin-based triple therapy improved eradication rates from 62.5% to 83.3%. R
DGL also promotes mucosal healing and ulcer repair independently of its anti-H. pylori effects, making it a logical inclusion in both eradication and repair phases.
Dose: 1.5-3g daily between meals.
Zinc Carnosine (Polaprezinc)
Zinc carnosine (the chelated combination of zinc and L-carnosine, known as polaprezinc) combines antimicrobial zinc with the mucosa-protective dipeptide carnosine.
In a randomized trial, adding polaprezinc to standard triple therapy (lansoprazole, amoxicillin, clarithromycin) significantly increased the H. pylori eradication rate compared to triple therapy alone, without increased toxicity. R
Zinc carnosine protects gastric mucosa, speeds tissue repair at ulceration sites, and enhances the local antimicrobial environment.
Dose: 75mg zinc carnosine twice daily with meals.
Vitamin C (Ascorbic Acid)
High concentrations of vitamin C in gastric juice directly inhibit H. pylori urease, the enzyme the organism uses to neutralize stomach acid for survival.
Without functional urease, H. pylori loses its primary acid-defense mechanism and becomes vulnerable.
An RCT found that adding 500mg vitamin C daily to standard quadruple therapy increased eradication from 48.8% to 78%. R
Dose: 500-1000mg daily during the eradication protocol.
Berberine
Berberine has documented anti-H. pylori activity through DNA intercalation (disrupting bacterial DNA replication) and broad antimicrobial effects.
Clinical trials adding berberine to pharmaceutical therapy report increased eradication rates and significant reduction in nausea and diarrhea side effects from antibiotics. R
Berberine also reduces H. pylori-driven gastric inflammation directly.
Dose: 500mg two to three times daily with meals.
Methylmethionine Sulfonium (Vitamin U)
Methylmethionine sulfonium (MMS), sometimes called "Vitamin U," is derived from methionine and found concentrated in raw cabbage.
It is not technically a vitamin but has 70+ years of human research supporting its role in healing peptic and duodenal ulcers through direct mucosal repair.
It repairs damaged and eroded intestinal mucosa, reduces gastric inflammation, and supports the mucosal layer that H. pylori degrades.
Dose: 200mg daily, or consume 1 cup of fresh raw cabbage juice daily (the original therapeutic vehicle used in early ulcer research).
Garlic (Allicin)
Garlic's active antimicrobial compound allicin (diallyl thiosulfinate) has documented in vitro bactericidal activity against H. pylori, including antibiotic-resistant strains, with inhibitory concentrations achievable in the gastric environment. R
A systematic review and meta-analysis of 8 RCTs found that allicin as an add-on to PPI triple therapy or bismuth quadruple therapy achieved an eradication rate of approximately 93.8% vs 83.6% in control groups (OR = 2.75, p < 0.001), with significantly better ulcer healing rates. R
The evidence for allicin-specific garlic products added to pharmaceutical therapy is stronger than for fresh garlic alone, because allicin content and bioavailability vary widely in commercial preparations.
Dose: Allicin-standardized garlic extract providing 2-5mg allicin daily, or 2-4 fresh crushed cloves (allicin is released by crushing and degrades within minutes).
Saccharomyces boulardii
Saccharomyces boulardii is a beneficial yeast with the most consistent probiotic evidence for H. pylori eradication.
A meta-analysis of 18 RCTs including 3,592 patients found that S. boulardii supplementation on standard eradication therapy significantly increased H. pylori eradication rates and reduced the incidence of total side effects including diarrhea, bloating, constipation, and nausea. R
A 2025 meta-analysis confirmed these findings, with the bismuth quadruple therapy plus S. boulardii arm achieving 93.18% eradication vs 65.2% for standard triple therapy alone in per-protocol analysis. R
S. boulardii also upregulates secretory IgA in the gut, stabilizes tight junctions of gastric mucosal epithelial cells, and reduces the abundance of antibiotic-resistance genes during treatment. R
Dose: 500mg twice daily during the eradication protocol and for 4 weeks after.
Mucosal Repair After Eradication
Eradicating H. pylori is only half the work.
The gastric mucosa, parietal cells, and intestinal barrier have all been damaged by months to years of infection and inflammation.
Repair is required.
DGL continues to be useful post-eradication for mucosal healing.
Zinc Carnosine: 75mg twice daily for 60-90 days post-eradication accelerates mucosal restoration.
L-Glutamine: the preferred fuel for intestinal epithelial cells; 5g twice daily supports barrier repair. R
Colostrum: contains growth factors (IGF-1, EGF, TGF-beta) that directly stimulate gastric mucosal regeneration. R
Probiotics: Lactobacillus acidophilus and Bifidobacterium longum alongside S. boulardii support microbiome reconstitution after eradication and antibiotic use.
Betaine HCl: once eradication is confirmed and significant gastric healing has occurred (typically 6-8 weeks post-treatment), restoring gastric acid with betaine hydrochloride supplements helps normalize B12, iron, and calcium absorption that hypochlorhydria had impaired.
Nutrient repletion: B12, iron, folate, magnesium, and calcium should all be assessed and supplemented as indicated given the documented depletion from H. pylori-associated hypochlorhydria.
What To Stay Away From
- NSAIDs (ibuprofen, naproxen, aspirin) during active H. pylori infection or treatment: NSAIDs damage the gastric mucosal barrier independently, and the combination of H. pylori and NSAID use dramatically increases peptic ulcer risk
- Alcohol: damages gastric mucosa, inhibits mucosal healing, and may reduce antibiotic efficacy during treatment
- High-salt diet: significantly exacerbates H. pylori virulence and accelerates the progression from gastritis to gastric cancer; high salt diet is an independent risk factor for gastric cancer, and H. pylori and high salt appear to act synergistically R
- Serology to confirm eradication: H. pylori IgG antibodies remain elevated for months to years after successful eradication; post-treatment testing must use the C13 urea breath test or stool H. pylori antigen test, not serology
- Proton pump inhibitors used long-term without addressing the underlying H. pylori: PPIs reduce symptoms without clearing the infection, and long-term PPI use itself causes hypochlorhydria with the same downstream nutrient depletion consequences as the infection itself; it is common for H. pylori to be missed because the patient's symptoms are suppressed by PPIs
- Relying on natural agents alone for CagA-positive, high-virulence infections: the risk profile of CagA-positive strains (gastric cancer, severe peptic ulcer disease) justifies the higher eradication rates achievable with pharmaceutical triple or quadruple therapy; natural protocols are appropriate adjuncts but should not substitute for pharmaceutical therapy in high-virulence presentations
- Treating the patient without treating household contacts: H. pylori transmits within families via oral-oral and fecal-oral routes; untreated household contacts are the most common source of reinfection; when one family member tests positive, screening other household members is clinically appropriate
Testing
First-Line Testing
PCR-based stool testing via the GI-MAP (Diagnostic Solutions) is the test of choice for H. pylori in functional medicine because it provides:
- Quantitative H. pylori load (not just positive/negative)
- All seven virulence factors (BabA, CagA, CagPAI, DupA, IceA, OipA, VacA) in a single result
- Comprehensive picture of the full gut ecosystem alongside the H. pylori result (dysbiosis, other pathogens, inflammatory markers, digestive function)
The Gut Zoomer (Vibrant Wellness) also detects H. pylori via multiplex PCR alongside full microbiome analysis, intestinal permeability markers, and gut immune function.
Conventional Testing Options
C13 urea breath test is the non-invasive gold standard for both initial diagnosis and post-treatment eradication confirmation.
The patient swallows C13-labeled urea; if H. pylori is present, its urease converts the labeled urea to C13-labeled CO2 that appears in exhaled breath.
Sensitivity and specificity are both above 95%.
This test must be performed at least 4 weeks after completing antibiotic therapy and at least 2 weeks after stopping PPIs to avoid false negatives.
Stool antigen test (HpSA) detects H. pylori proteins directly in stool via enzyme immunoassay.
Sensitivity is approximately 94% and specificity 97% for initial diagnosis.
Also appropriate for post-treatment confirmation.
Endoscopy with biopsy: rapid urease test (CLO test) on biopsy tissue, histology, and culture from endoscopy.
Culture allows antibiotic sensitivity testing, which is increasingly valuable given rising clarithromycin and metronidazole resistance rates.
Serology (H. pylori IgG): useful for initial epidemiological screening but not for confirming eradication; antibodies persist for months to years after clearance.
Downstream Consequence Testing
When H. pylori is confirmed, always assess:
CBC with Differential: anemia pattern (hypochromic microcytic for iron deficiency, macrocytic for B12 deficiency), neutropenia as a sign of concurrent infection burden.
Vitamin B12 and Folate: both commonly depleted by H. pylori-associated hypochlorhydria.
Iron Studies + Ferritin: ferritin is the most sensitive marker of iron stores; low ferritin in the presence of H. pylori is a combination that confirms iron-deficiency contribution from the infection.
Nutrient Zoomer (Vibrant Wellness): most comprehensive option if a full micronutrient picture is needed, covering B12, folate, iron, magnesium, calcium, vitamin C, and zinc alongside full vitamin and mineral panel.
Foundation Zoomer (Vibrant Wellness): baseline liver function, CBC, CMP, and iron studies in a single panel.
Mechanisms Of Action
Simple:
- H. pylori produces urease to neutralize stomach acid locally, allowing it to survive in an environment designed to kill bacteria, then burrows into the protective mucus layer of the stomach where immune cells cannot reach it.
- By damaging the acid-producing parietal cells, H. pylori progressively reduces stomach acid, which in turn blocks the absorption of vitamin B12, iron, calcium, magnesium, and folate because all of these require gastric acid for their release or solubility.
- The bacteria injects a toxin called CagA directly into stomach cells, hijacking their signaling pathways and driving uncontrolled cell growth that, over decades, can progress to gastric cancer.
- The chronic inflammation from H. pylori degrades the tight junctions between intestinal cells, creating leaky gut that allows food antigens into the bloodstream and triggers food sensitivities, skin problems, and autoimmune reactions.
- Mastic gum kills H. pylori by disrupting its cell membrane through its acidic resin fractions; vitamin C neutralizes H. pylori urease, stripping away its acid-defense shield; zinc carnosine supports mucosal repair while zinc provides a direct antimicrobial environment.
Advanced:
- Urease mechanism and ammonium buffering: H. pylori urease is a nickel-containing multi-subunit enzyme (UreAB) with extraordinary catalytic capacity, producing enough ammonia to buffer the immediate microenvironment to pH 6-7 even in a pH 2 stomach. H. pylori also expresses UreI, an inner membrane proton-gated urea channel that opens only at low pH, ensuring ammonia production precisely when acid exposure increases. The resulting ammonium ions are directly toxic to gastric epithelial cells (contributing to mucosal damage) while simultaneously protecting the bacteria. R
- CagA T4SS injection and oncogenesis: The Cag pathogenicity island (Cag PAI) encodes a type IV secretion system (T4SS), a molecular syringe that contacts the host cell surface and directly injects CagA protein into the cytoplasm. Once inside, CagA is phosphorylated at EPIYA motifs by Src and Abl kinases. Phosphorylated CagA binds and activates SHP2 phosphatase, which drives sustained ERK activation and the "hummingbird phenotype" (elongated cell morphology, increased motility). Unphosphorylated CagA disrupts E-cadherin-beta-catenin complexes, activating Wnt/beta-catenin transcription and driving proliferative gene expression. CagA also activates NF-kB through interaction with TAK1 and LUBAC, and disrupts PAR1 polarity kinase, causing loss of epithelial architecture. Collectively, these effects simultaneously promote inflammation, cell proliferation, and loss of tumor suppressor function. R
- Iron competition and hepcidin modulation: H. pylori competes directly for host iron, which it requires for growth and virulence gene expression. It expresses lactoferrin-binding proteins (HopQ, AlpAB) and produces siderophores to scavenge iron from the gastric environment. In parallel, H. pylori infection stimulates liver hepcidin production through IL-6-dependent JAK-STAT3 signaling. Hepcidin causes ferroportin internalization and degradation on macrophages, hepatocytes, and enterocytes, trapping iron intracellularly and reducing serum iron availability. This creates functional iron deficiency through two simultaneous mechanisms: impaired dietary iron absorption from hypochlorhydria, and hepcidin-mediated restriction of iron release from stores. R
- Mastic gum mechanism: The active anti-H. pylori fraction of mastic gum is its acidic resin components, particularly isomasticadienolic acid and oleanolic acid. These triterpenic acids disrupt H. pylori cell membrane integrity through detergent-like insertion into the lipid bilayer, causing cell morphology abnormalities (blebbing, cellular fragmentation) visible by electron microscopy at sub-bactericidal concentrations. They also inhibit DNA gyrase (topoisomerase II) in H. pylori, blocking DNA replication. Notably, mastic gum appears active against metronidazole-resistant and clarithromycin-resistant strains, making it a meaningful adjunct when antibiotic resistance reduces pharmaceutical efficacy. R
- Vitamin C and urease inhibition: Ascorbic acid at concentrations achievable in gastric juice (physiological intragastric vitamin C is approximately 100-250 mcmol/L in healthy individuals but suppressed to near zero in H. pylori infection) directly inhibits H. pylori urease activity through oxidation of the essential cysteine residue in the active site. Without functional urease, H. pylori cannot buffer pericosmic pH, loses its acid resistance, and cannot maintain colonization in the gastric antrum. Vitamin C also stimulates gastric mucus production and mucosal immunity. The direct suppression of gastric vitamin C by H. pylori is itself a virulence mechanism that removes this natural chemical defense. R
More Research
- H. pylori is carried by approximately 50% of the global population and causes over 90% of duodenal ulcers and approximately 80% of gastric ulcers; it is classified as a Group 1 definite carcinogen by IARC and is the primary modifiable cause of gastric adenocarcinoma. R
- Virulence factor testing via the GI-MAP changes clinical decision-making: a CagA-positive, VacA-positive infection carries substantially higher risk for peptic ulcers and gastric cancer than a virulence-factor-negative infection, and the urgency and aggressiveness of eradication should reflect this stratification.
- Iron-deficiency anemia that fails to respond to iron supplementation should prompt H. pylori testing even in the absence of gastrointestinal symptoms; the combination of iron deficiency and H. pylori is common and the anemia will not resolve while the infection persists. R
- Post-treatment confirmation of eradication is not optional: a meaningful proportion of patients who complete a full antibiotic course fail to eradicate H. pylori due to resistance, non-compliance, or incomplete bacterial killing; the C13 urea breath test at 4-6 weeks post-treatment is the standard confirmation method. R
- Adding mastic gum, DGL, berberine, zinc carnosine, vitamin C, allicin (garlic), and/or Saccharomyces boulardii to pharmaceutical triple therapy consistently improves eradication rates and reduces antibiotic-related side effects across multiple RCTs; a meta-analysis of allicin specifically found an eradication rate of 93.8% vs 83.6% in the control arm (OR = 2.75, p < 0.001), and a meta-analysis of S. boulardii found significantly improved eradication rates and reduced side effects across 18 RCTs with 3,592 patients. R R
- H. pylori eradication is a first-line intervention for H. pylori-associated ITP: the molecular mimicry mechanism between CagA and platelet surface glycoproteins drives anti-platelet autoantibody production, and eradication removes the antigen source; long-term follow-up studies show platelet recovery sustained 7 or more years post-eradication in responding patients. R
- A meta-analysis confirmed that H. pylori infection correlates with both Graves' disease and Hashimoto's thyroiditis, and that H. pylori eradication reduces thyroid autoantibodies; CagA-positive strains carry the strongest association with thyroid autoimmunity through molecular mimicry with thyroperoxidase. R
- The Correa cascade from H. pylori infection to gastric cancer is well-characterized and takes decades, but eradication at any point in the non-atrophic or early atrophic gastritis stage can halt or reverse the progression; once intestinal metaplasia is established, cancer risk persists even after eradication, requiring endoscopic surveillance. R
- Household contact screening is clinically appropriate after any confirmed H. pylori diagnosis; reinfection from an untreated household member is a common reason eradication protocols fail to prevent recurrence.
- For comprehensive gut assessment when H. pylori is suspected, the GI-MAP provides H. pylori quantification, full virulence factor panel, co-pathogen screening, dysbiosis mapping, and inflammatory markers in one result, making it the most clinically informative single test available.
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Electrolyte Complex
1 scoop/day
CoQ10
200mg/day
Magnesium Glycinate
400mg at bedtime