The Hypermobility-MCAS-POTS Triad: Why hEDS, Mast Cells, and Dysautonomia Travel Together
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.
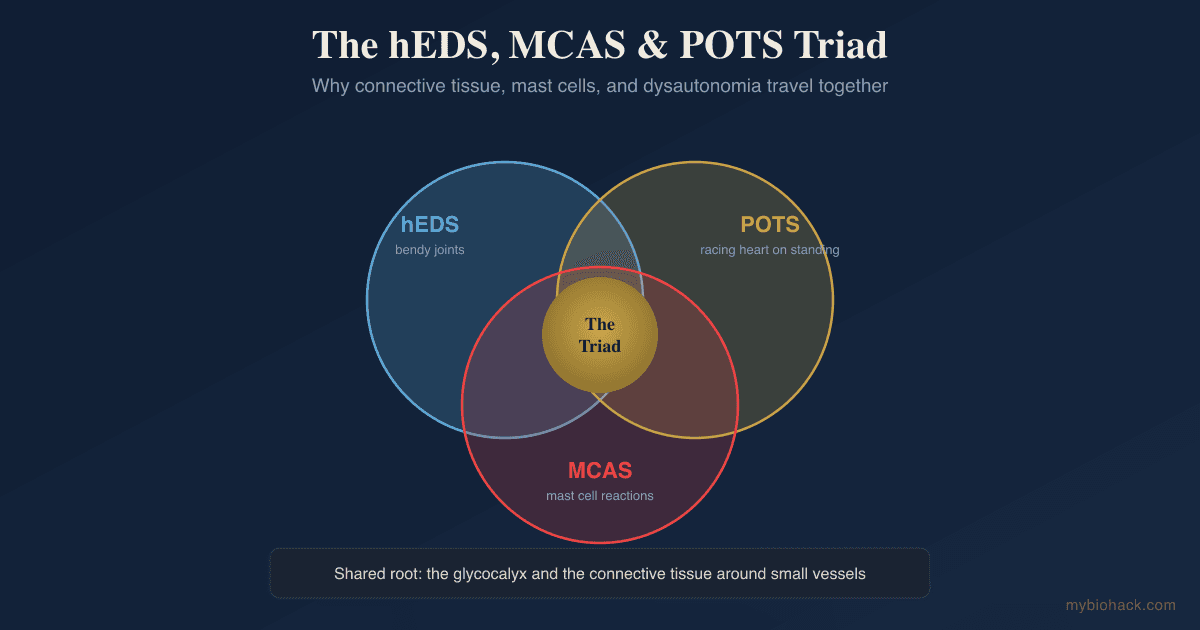
Three conditions keep showing up in the same patients: bendy joints, a racing heart on standing, and a body that reacts to foods, scents, and temperature like an allergic emergency.
In this post, we will discuss what the triad is, why these three diagnoses cluster so tightly, the mechanism that ties connective tissue to blood vessels and mast cells, how it overlaps with post-viral illness, the protocol I use to stabilize it, what to avoid, how to test it, and the genetics behind it.

Basics Of The Triad
The triad is the frequent co-occurrence of three conditions in the same person.
The first is hypermobile Ehlers-Danlos Syndrome (hEDS), a heritable connective tissue disorder defined by generalized joint hypermobility, joint instability, soft skin, and chronic musculoskeletal pain. R
The second is Mast Cell Activation Syndrome (MCAS), a condition where mast cells release their mediators inappropriately, producing flushing, hives, abdominal pain, food and chemical reactions, and sometimes anaphylaxis.
The third is Postural Orthostatic Tachycardia Syndrome (POTS), a form of dysautonomia defined by a sustained heart rate rise of at least 30 beats per minute within ten minutes of standing, without a drop in blood pressure.
Each one is its own diagnosis, but they are found together far more often than chance would predict.
In one cohort of patients with hypermobility, more than half reported physician-diagnosed POTS and roughly a third reported MCAS, with about one in four carrying the full clinical triad of hypermobility, POTS, and mast cell disease. R
Surveys of hEDS patients consistently rank POTS, migraine, irritable bowel syndrome, and anxiety among the most common co-diagnoses. R
I want to be honest at the outset that the strength of this clustering is contested.
A 2025 multicenter retrospective review argued that MCAS is not statistically associated with hEDS or POTS once you apply strict consensus criteria, and a separate appraisal questioned whether the genetic link to mast cells holds up. R R
There is a big MAYBE here, and I will return to it.
What is not contested is that a real subset of patients clearly has all three, and that subset needs a framework that explains the overlap rather than three separate specialists who never talk to each other.
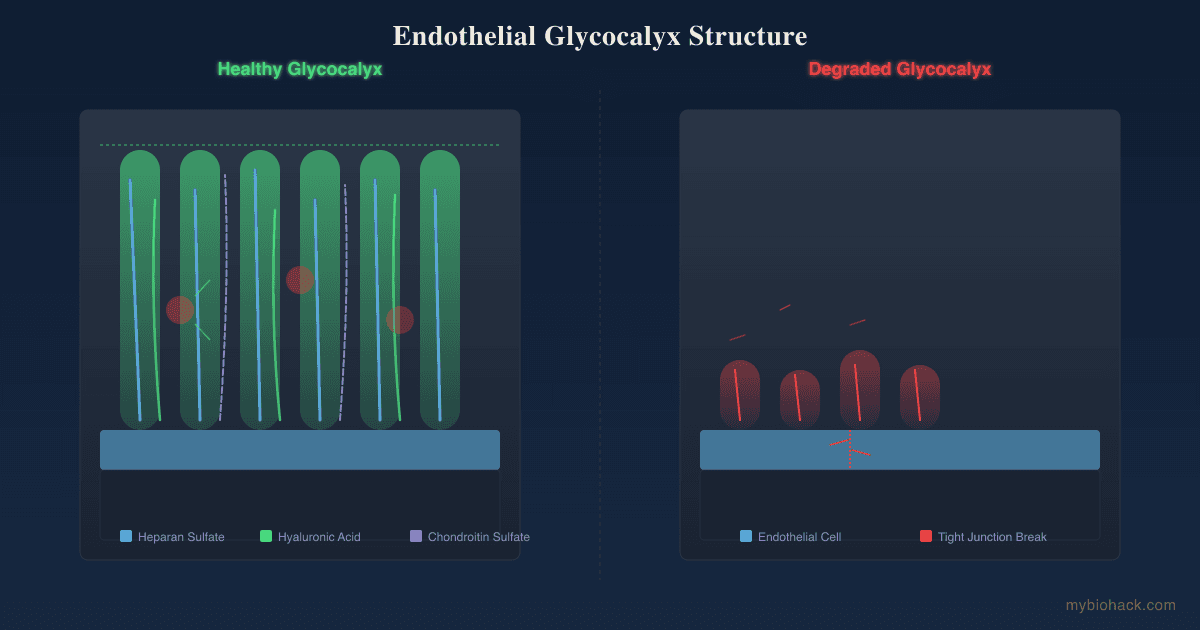
That framework, in my model, is Junction Dysfunction (JD), the umbrella pathology that develops when the glycocalyx (the sulfated sugar coating on the inside of blood vessels) breaks down.
What Causes The Connection
The mainstream explanation is the simplest place to start.
All three conditions share connective tissue as the common substrate.
Collagen is the scaffold of joints, skin, blood vessel walls, and the matrix that mast cells live in, so a defect in connective tissue can plausibly touch all three systems at once.
In hEDS the collagen defect makes ligaments lax, which is what you feel as hypermobility.
The same laxity affects the walls of veins, which is the leading mechanical explanation for POTS in this population.
Veins in hypermobile patients distend more than they should under ordinary standing pressure, blood pools in the legs and abdomen, less blood returns to the heart, and the heart compensates by beating faster. R R
Mast cells enter the picture because they physically reside in connective tissue, packed in a perivascular position right against the small vessels. R
That puts them at the exact interface where connective tissue laxity, blood pooling, and low tissue perfusion all happen.
There is also a genetic thread that some researchers think runs through all three: Hereditary Alpha-Tryptasemia (HαT), an inherited increase in the copy number of the alpha-tryptase gene that raises baseline tryptase and is more common in people with hypermobility and autonomic symptoms. R R
My framing adds a layer underneath all of this.
I think the deepest shared node is not collagen alone but the glycocalyx and the small vessels it protects, because that is the one structure that is simultaneously a connective tissue surface, the regulator of vascular leak, and the home address of the mast cell.
When the glycocalyx degrades, you get fluid leak, blood pooling, and mast cell provocation in the same place at the same time, which is the cluster the triad describes.
I cover the connective tissue side of this in depth in the JD chapter Why Do I Have A Connective Tissue Disorder All Of A Sudden?
How The Connective Tissue Defect Drives Each Condition
To see why the three travel together, follow the connective tissue defect into each system.

The Vessel Wall And POTS
Blood vessels are mostly collagen, and veins are thin-walled, low-pressure structures that depend on wall tone to push blood back uphill against gravity.
When the collagen and the glycocalyx that lines those vessels are compromised, two things happen.
First, the veins become too compliant and pool blood, the classic connective tissue explanation for orthostatic intolerance. R
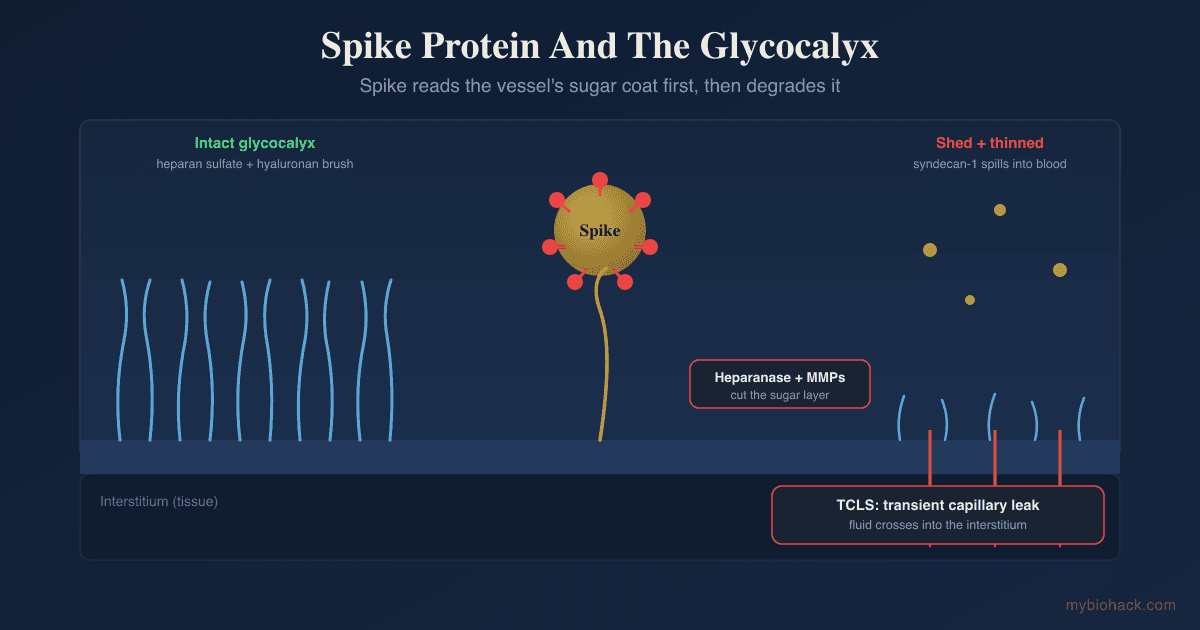
Second, and this is my hypothesis, the microcapillary bed loses functional channels through Transient Capillary Leak Syndrome (TCLS), a term I coined for the micro-level leak that drains fluid from the vessel into the tissue.
I call the resulting loss of vascular adaptability Vaso-Adaptive Disorder (VAD) rather than "vascular POTS," because the core problem is a loss of the body's ability to adapt to vasoactive stress like standing, heat, and exertion.
When adrenergic activation then recruits platelets and fibrin into those same leaky microcapillaries, you get the micro-clotting variant I call Adrenergic-Based Vaso-Adaptive Disorder (ABVAD) rather than "hyperadrenergic POTS."
I work through how this raises heart rate, and how it explains the renin-aldosterone paradox in POTS, in microcapillaries and vascular POTS and blood clots, platelets, and fibrin.
The full vascular argument is in the JD chapters Microcapillaries And Vascular POTS and Blood Clots, Platelets, Fibrin, And Autonomic POTS, and I keep a more general overview in Root Causes Of POTS.
The Mast Cell And MCAS
Mast cells sit in connective tissue, perivascularly, and one of their jobs is to release histamine, a vasodilator, in regions that are low on oxygen.
In my model the connective tissue patient has less capillary reserve to begin with, so tissue beds go hypoxic more easily, and the mast cells in those beds degranulate trying to re-open the vasculature.
That makes the connective tissue defect a direct provocation for mast cell activation rather than a coincidence.
Histamine then loosens endothelial junctions further, which worsens the leak that started the cycle, so the mast cell both responds to and amplifies the vascular problem.
When the glycocalyx is shedding, the enzymes immune cells use to cut through it, hyaluronidase and matrix metalloproteinases, also degrade the surrounding connective tissue, which is collateral damage that further weakens the same collagen the patient was already short on.
I lay out the hypoxia-to-mast-cell logic in the JD chapter Mitochondria And Mast Cells In Hypoxia, and the upstream drivers of histamine load in Why Histamine Produces So Many Symptoms.
The Nerve-Mast Cell Loop
The third connection is neurogenic.
Mast cells communicate with sensory nerves, and substance P released from nerves drives mast cells to degranulate, which then sensitizes the nerves further.
In a dysautonomic patient with sympathetic dominance, this loop runs hot, which is part of why MCAS and POTS symptoms flare together rather than separately.
I cover this nerve-to-mast-cell circuit in Mast Cells, Substance P, And Neurogenic Inflammation.
Overlapping Conditions
The triad rarely arrives alone.
The conditions that overlap most often, in alphabetical order:
- Chronic fatigue and ME/CFS (post-exertional malaise overlaps heavily with the POTS population) R
- Gastrointestinal dysmotility and IBS (the gut is a connective tissue and mast cell organ, and dysmotility tracks with hEDS) R
- Histamine intolerance (low DAO enzyme activity stacks on top of mast cell mediator load)
- Long COVID and post-viral illness (spike protein and chronic immune activation degrade the glycocalyx and can unmask the triad)
- Migraine (neurovascular and mast cell driven, one of the most common co-diagnoses) R
The long COVID overlap is the one I watch most closely.
A post-viral hit can degrade the glycocalyx and push a previously compensated hypermobile person into full triad expression, which is why a lot of "new onset POTS and MCAS after a virus" stories are really the triad being unmasked.
I walk through the post-viral version of this in 7 Steps To Naturally Treat Long COVID and the underlying barrier biology in The Glycocalyx.
How To Improve The Triad
The logic of the protocol is to stabilize the three loops at once: hold blood volume up so the heart does not have to race, calm the mast cell so it stops amplifying leak, and rebuild the connective tissue and glycocalyx that protect the vessels.
Nonpharmacological measures are the foundation, and the few that have been tested directly are salt, fluids, compression, and graded exercise. R
1. Raise Blood Volume With Salt And Fluids
Increasing sodium and water is the first-line intervention for the POTS component because it expands plasma volume and reduces the reflex tachycardia.
Aim for generous fluids through the day and add electrolytes rather than plain water, since plain water without sodium will not hold volume.
Electrolytes:
A high-sodium electrolyte mix is the simplest way to hit the salt target without forcing yourself to eat salt straight.
2. Use Compression
Abdominal and lower-body compression reduces venous pooling, and in a randomized crossover tilt study it lowered heart rate in a dose-dependent way, with abdominal compression outperforming leg-only compression. R
Compression stockings:
Waist-high or abdominal compression at 20-30 mmHg gives the most benefit for the pooling that drives the tachycardia.
3. Stabilize Mast Cells
The goal is to make mast cells harder to trigger and to soak up the histamine they release.
Quercetin:
Quercetin blocked human mast cell release of histamine, tryptase, and inflammatory cytokines more effectively than cromolyn in human studies, and it works by stabilizing the cell membrane and down-regulating the IgE receptor. R R
Luteolin:
Luteolin is a closely related flavonoid that stabilizes mast cells and crosses into the brain, useful when neuroinflammation and brain fog are part of the picture.
Palmitoylethanolamide:
PEA calms mast cell and glial activation and pairs well with luteolin for the nerve-mast cell loop.
Vitamin C:
Vitamin C helps degrade histamine and supports the cofactor pool the vessels need.
4. Lower The Histamine Load
If reactions track with food, reduce the histamine coming in while you stabilize the cells making it.
DAO enzyme:
Diamine oxidase taken before meals breaks down dietary histamine in the gut, which lightens the total mediator load.
A lower-histamine diet during the stabilization phase reduces flares while you rebuild, and the full approach is in Why Histamine Produces So Many Symptoms.
5. Support Connective Tissue And The Glycocalyx
This is the layer mainstream triad protocols skip, and in my model it is the one that actually addresses the shared root.
Vitamin C is also the rate-limiting cofactor for collagen cross-linking, so it does double duty here.
Magnesium:
Magnesium glycinate supports vascular tone, calms the adrenergic overdrive of ABVAD, and the glycine portion supplies a key collagen amino acid.
Sulfated polysaccharides such as fucoidans help rebuild the glycocalyx itself, and I make the case for why glycocalyx repair belongs in any triad protocol in Rebuilding Trees Of The Forest: Healing The Glycocalyx.
6. Graded, Recumbent Exercise
Exercise is protective for POTS but only if it starts horizontal and builds slowly, because upright exertion early on provokes both the tachycardia and mast cell degranulation.
Recumbent bike, rowing, and swimming let you build cardiovascular conditioning without standing, and strength work for the legs improves the muscle pump that fights pooling.
This has to be graded, because overexertion damages the glycocalyx and triggers post-exertional crashes in this population.
7. Address The Nervous System
Sympathetic dominance ties the whole triad together, so calming the autonomic nervous system lowers all three loops.
Vagal tone, breathwork, and limbic retraining matter here, because I do not see people fully stabilize on supplements and compression alone without working on the nervous system driving the flares.
The acetylcholine and vagal side of this is in Nicotinic Acetylcholine Receptors.
What To Stay Away From
Some common interventions make the triad worse.
- High-histamine and fermented foods during the unstable phase (aged cheese, cured meat, alcohol, kombucha, leftovers) push the mast cell load higher
- L-arginine and nitric oxide boosters can worsen oxidative leak in a degraded glycocalyx and reactivate latent viruses, so I avoid them in this population
- Prolonged standing and upright heat exposure like hot showers and saunas early on provoke pooling and degranulation before the system is stable
- Sudden upright high-intensity exercise triggers tachycardia and post-exertional crashes, which is why graded recumbent work comes first
- Sulfite-heavy foods and supplements (wine, dried fruit, some preservatives) provoke mast cells in people who do not clear sulfites well
That last one is worth a note, because in my clinical experience sulfite-sensitive patients almost always have trouble converting sulfite to sulfate, which ties directly into the sulfated glycocalyx, and I cover that link in What You Need To Know About Sulfur.
Testing
The goal of testing is to confirm each leg of the triad objectively and to look for the shared root underneath.
POTS And Autonomic Testing
POTS is confirmed with a tilt table test or a ten-minute active stand test, documenting a sustained heart rate rise of at least 30 beats per minute on standing without a fall in blood pressure.
For the broader autonomic and adrenal picture, I use the POTS / Dysautonomia panel (Vibrant Wellness) to map catecholamine metabolites, cortisol rhythm, adrenal output, and adrenergic receptor autoantibodies.
Mast Cell Markers
The single most useful blood marker is baseline tryptase, which also screens for the HαT genetics.
A baseline level above 11.4 ng/mL is generally considered elevated, and the consensus way to prove acute mast cell activation is to draw tryptase during a flare and confirm it rises above the patient's baseline times 1.2 plus 2 ng/mL. R R
I use the Tryptase test (Quest Diagnostics) for the baseline value and the Plasma Histamine test (Quest Diagnostics) alongside it.
Other supportive markers include 24-hour urinary N-methylhistamine and prostaglandin metabolites, which are harder to collect but more specific to mast cell release.
Autoimmune And Neural Markers
Because dysautonomia in this group is often accompanied by adrenergic and muscarinic receptor antibodies, the Immune Zoomer (Vibrant Wellness) maps systemic autoantibodies and mast cell related immune reactivity, and the Neural Zoomer (Vibrant Wellness) screens autonomic and blood-brain-barrier autoimmunity.
Oxidative Stress And Mitochondrial Function
To see the perfusion and redox layer that drives the hypoxia-mast cell loop, I use the Cellular Zoomer (Vibrant Wellness) for organic acids, mitochondrial function, and oxidative stress markers.
Genetic Confirmation
hEDS itself has no genetic test, so it remains a clinical diagnosis made with the Beighton score and the 2017 criteria.
The other EDS subtypes do have genetic tests, and for the mast cell genetics, TPSAB1 copy number analysis for HαT is run by specialized labs such as ARUP via droplet digital PCR.
For the broader detox and sulfur genetics I look at, the Toxin Genetics panel covers the relevant variants.
If you want help interpreting these together rather than as isolated results, this is the kind of multi-system picture I work through on a consultation.
Mechanisms Of Action
Simple:
- Weak connective tissue means stretchy joints, floppy veins that let blood pool when you stand, and a fragile matrix where mast cells sit, so one defect can cause loose joints, a racing heart, and allergic-type reactions at the same time.
- Mast cells live right next to small blood vessels in low-oxygen connective tissue, so when those tissues run short on blood, the mast cells dump histamine trying to open the vessels, which then leak more and feed the cycle.
Advanced:
- Venous compliance and orthostatic tachycardia. Defective collagen in vein walls increases venous distensibility, so upright hydrostatic pressure causes excessive dependent pooling, reduced venous return, and a compensatory sympathetically driven rise in heart rate, which is the mechanical model of POTS in the hypermobile population. R
- Glycocalyx shedding and TCLS. In my model, degradation of the sulfated endothelial glycocalyx via hyaluronidase, heparanase, matrix metalloproteinases, and reactive oxygen species opens tight junctions and produces microvascular leak, draining plasma into the interstitium, which I develop into the full VAD mechanism in microcapillaries and vascular POTS.
- Perivascular mast cell provocation. Mast cells are concentrated perivascularly in connective tissue, where reduced capillary reserve creates local hypoxia that promotes degranulation, and the released histamine, tryptase, and proteases increase vascular permeability and degrade matrix further, coupling the mast cell loop to the vascular leak loop. R
- Hereditary alpha-tryptasemia. Increased germline copy number of the alpha-tryptase gene TPSAB1 raises basal tryptase in a gene-dose manner and is enriched in cohorts with hypermobility and dysautonomia, providing one inherited mechanism that could raise mast cell reactivity across the triad, though its causal weight is debated. R R
Genetics
TPSAB1
TPSAB1 encodes alpha-tryptase, the protease stored in mast cell granules.
Extra germline copies of the alpha-tryptase allele raise baseline serum tryptase and define Hereditary Alpha-Tryptasemia, which affects roughly 5 to 7 percent of Western populations.
Copy number above the normal range correlates in a gene-dose fashion with higher tryptase and greater symptom severity, and HαT is over-represented in people who also have hypermobility and autonomic symptoms. R
COL5A1 And COL5A2
These genes encode type V collagen, which controls how type I collagen fibrils assemble in skin, tendon, and vessel wall.
Loss-of-function variants are the main cause of classical EDS, producing fragile, hyperextensible skin and joint hypermobility. R
COL3A1
COL3A1 encodes type III collagen, which is a major structural component of blood vessel and hollow organ walls.
Pathogenic variants cause vascular EDS, the most dangerous subtype, because the same collagen that gives arteries and the gut their tensile strength is defective, raising the risk of arterial and organ rupture. R
The hEDS Gene Is Still Unknown
Of the thirteen EDS subtypes defined in the 2017 international classification, twelve have an identified molecular cause, and hypermobile EDS is the exception. R
No single gene has been confirmed for hEDS, which is why it remains a clinical diagnosis based on the Beighton score, systemic features, and family history rather than a genetic test.
This is the central unsolved problem of the field, and it is the reason hEDS, the most common subtype, is also the least understood.
SUOX
SUOX encodes sulfite oxidase, the enzyme that converts sulfite to sulfate.
Reduced activity lets sulfites accumulate, which can provoke mast cell degranulation, and because sulfate is needed to build the sulfated glycocalyx, poor sulfite handling ties the mast cell and vascular legs of the triad together in my model.
rs7297662 is a variant associated with sulfite handling that I check in patients with strong sulfite reactivity.
More Research
The honest state of the evidence is that the co-occurrence is real in a defined subset but the causal links are still being argued.
A 2025 multicenter retrospective review found no statistical association between MCAS and either hEDS or POTS once strict consensus criteria were applied, and a separate appraisal argued the HαT link to hypermobility and dysautonomia is weaker than commonly claimed. R R
I take this seriously, because part of the triad's popularity comes from loose diagnosis, and applying real criteria to all three matters.
Drug evidence for the POTS leg is thin, with the best randomized data supporting propranolol and ivabradine for rate control, both of which are rate-limiting band-aids rather than root-cause fixes. R
For the renin-angiotensin and vascular side, I find blocking the AT1 receptor more interesting mechanistically than blunting heart rate, and I cover that logic in The Benefits Of Losartan.
Gastrointestinal dysmotility is an underappreciated fourth leg of this cluster and tracks tightly with hEDS, so a triad workup that ignores the gut misses a major driver of the mast cell load. R
My own working hypothesis, which I label as a hypothesis and not settled science, is that the glycocalyx and the microvascular leak of TCLS sit underneath all three legs, which would explain why post-viral hits that shed the glycocalyx so often unmask the full triad, and why glycocalyx repair belongs in the protocol even though no trial has tested that idea directly.
For biomarker tracking over time I use the POTS / Dysautonomia panel and the Immune Zoomer to watch the autonomic and mast cell markers move together as the protocol takes hold.
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Electrolyte Complex
1 scoop/day
CoQ10
200mg/day
Magnesium Glycinate
400mg at bedtime