NAFLD and NASH - What It Is and How to Treat It
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.
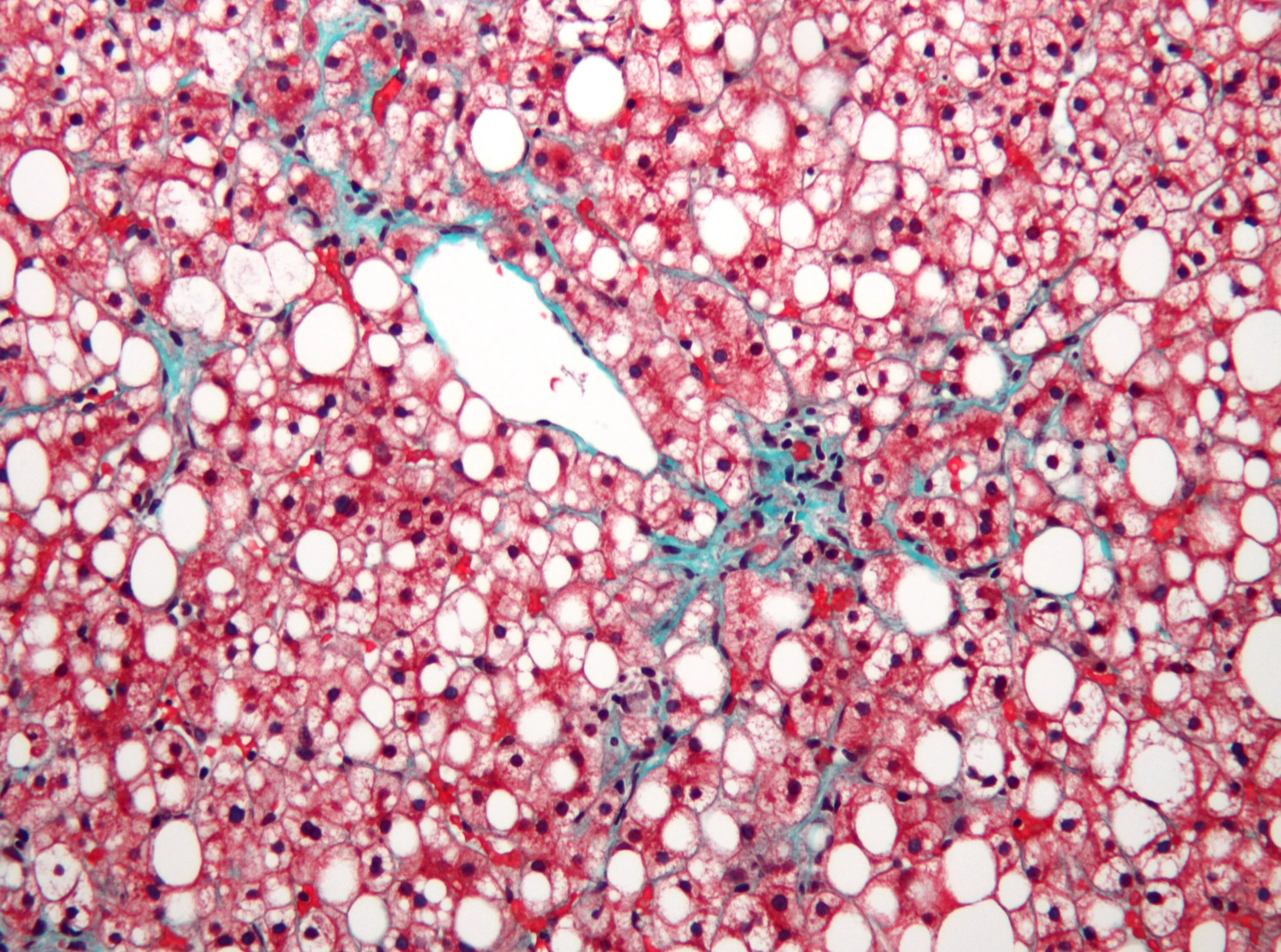
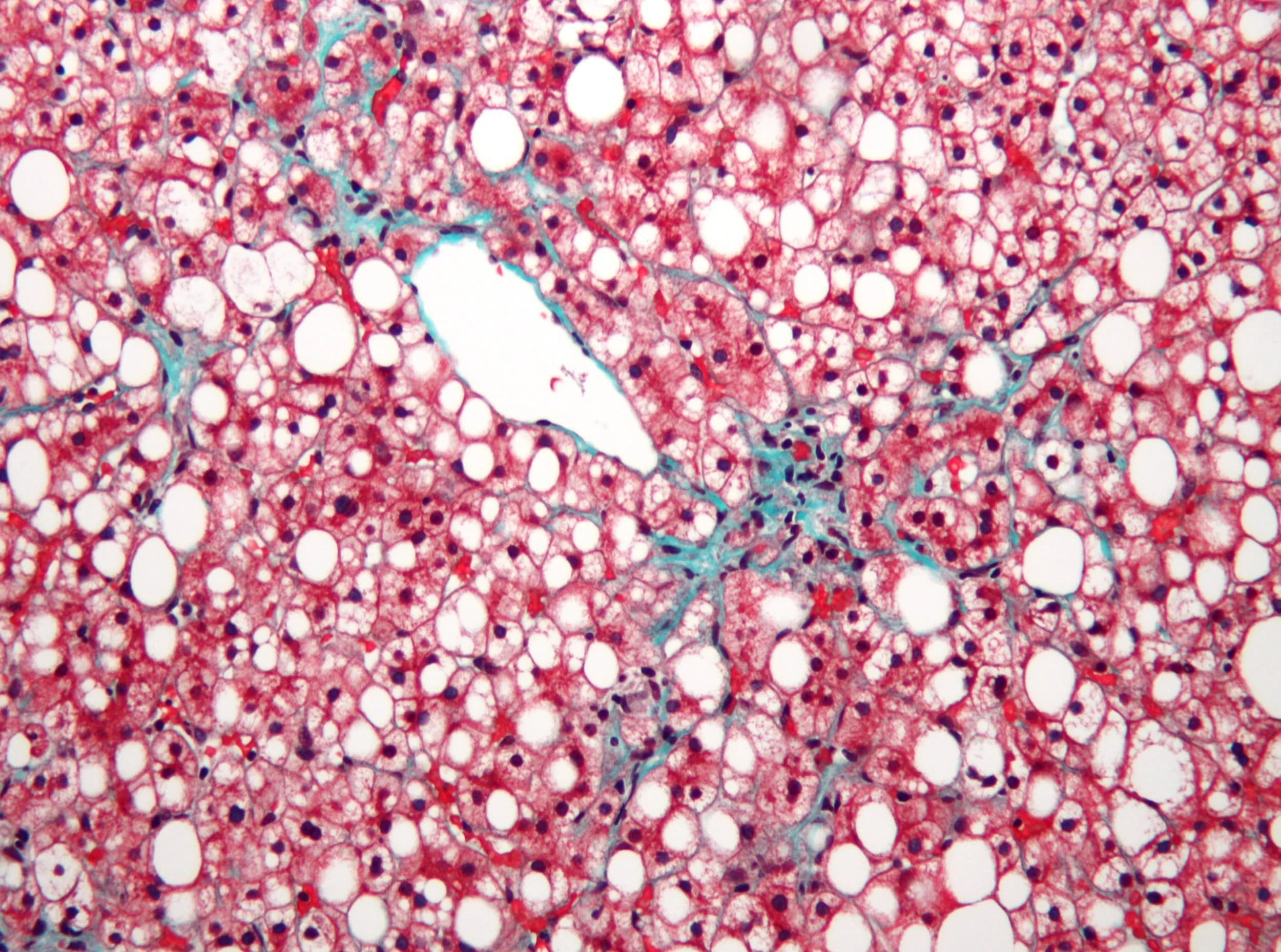
Micrograph of non-alcoholic fatty liver disease, demonstrating marked steatosis (fatty liver appears white).
Basics

It is estimated that at least 25% of American adults have some form of NAFLD, with about 6% of the general adult population having NASH and 2%–3% having NAFLD-related cirrhosis. R
Non-alcoholic steatohepatitis (NASH) is the most extreme form of NAFLD, and is regarded as a major cause of cirrhosisof the liver of unknown cause. R
Most people have a good outcome if the condition is caught in its early stages. R
To break this down simply:
The microbiome may get dysregulated.
Genetics, diet (including drugs and alcohol), lifestyle, and pathogens (amongst other complications) can influence a dysregulation.
This dysregulation can cause inflammatory responses, releasing cytokine, interluekin, and chemokine responses.
Microbiome
Cirrhosis and impaired liver function are associated with intestinal bacterial overgrowth, small bowel dysmotility, increased gut permeability, and decreased immunological defenses. R
DNA sequencing have allowed for more sophisticated analysis and sampling of the gut microbiota by culture-independent methods. R
Dysbiosis has been implicated in chronic metabolic disorders such as: R
Obesity
Metabolic syndrome R
Diabetes
Cardiovascular diseases
Patients with NASH have a significantly higher percentage of Clostridium coccoides than patients with NAFLD. R
Escherichia is the significantly disproportionate between obese children and pediatric patients with NASH. R
SIBO

Patients with obesity or NAFLD have a higher prevalence small intestinal bacterial overgrowth (SIBO). R R
Gut permeability and bacterial overgrowth correlate with severity of steatosis (not fibrosis or inflammation). R
There is seen reductions in: R
Lactobacillus
Pediococcus
Leuconostoc
Lactococcus
Alcoholic Liver Disease

Bacteroidaceae is lower in alcoholic patients than from non-alcoholic individuals. R
Administration of Lactobacillus reduced features of alcoholic liver disease, in animal models. R R R
Cirrhosis
Liver fibrosis may result cirrhosis, which can disrupt metabolic functions of the liver. R
Most patients with cirrhosis have intestinal bacterial overgrowth (more pathogenic than beneficial bacteria). R R R
Patients with low IGF-1 (insulin like growth factor) have advanced liver cirrhosis. R
Technical
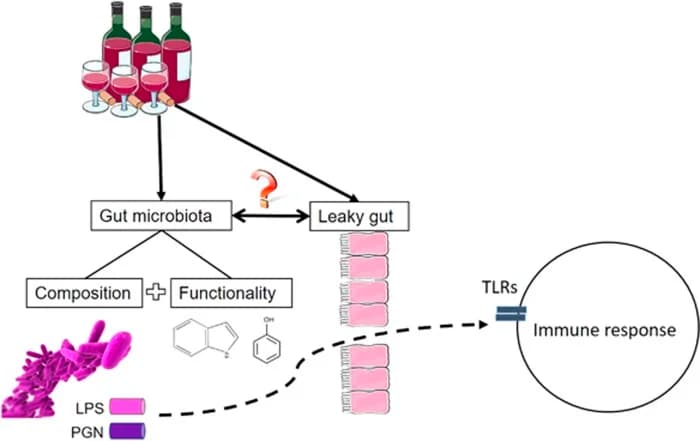
Intestinal inflammation might cause intestinal leakage and translocation of microbial products. Obesity is accompanied by inflammation in the colorectal mucosa; in obese individuals, diet-induced weight loss reduces colorectal inflammation and alters expression of inflammatory and cancer-related genes. R
In mice:
High-fat diets increase activity of the transcription factor NFκB and expression of tumor necrosis factor (TNF)α in the small intestine. Intestinal inflammation depends on the enteric microbiota; germ-free mice are protected from inflammation of the small intestine. R
More direct evidence for dysbiosis-induced intestinal inflammation and bacterial translocation has come from studies of mice deficient for Nlrp3 and Nlrp6. These mice cannot form cytoplasmic multi-protein complexes composed of nucleotide-binding domain and leucine-rich repeat containing proteins (NLRPs), called inflammasomes. Inflammasomes are sensors of exogenous PAMPs or endogenous damage-associated molecular patterns (DAMPs) that regulate cleavage of precursors to inflammatory cytokines such as pro-interleukin (IL)1β and pro-IL18. R
Loss of Nlrp3 and Nlrp6 inflammasomes is associated with intestinal dysbiosis and eventual inflammation of the colon, via the chemokine CCL5. Dysbiosis is characterized by an increase in Prevotella. R
Non Technical
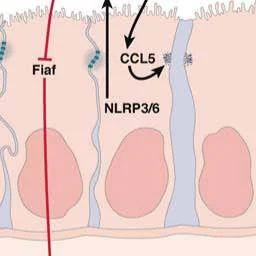
Effects of the Intestinal Microbiota on NAFLD and Progression to Steatohepatitis R
Diet- and host-induced changes in the intestinal microbiota contribute to the onset of NAFLD and NASH.
Bacterial changes can affect the liver and bacterial metabolites.
Direct host–microbiota interactions in the intestine alter intestinal homeostasis, which affects the liver.
Functional Consequences
Metabolites
Several changes in bacterial metabolites have been associated with obesity, and in particular, with NAFLD.
One of these metabolites is ethanol, a product of the intestinal microbiome.
Obese animals have higher blood concentrations of ethanol, determined by breath tests, than lean animals. R
Alcohol is absorbed and reaches the liver via the portal blood.
Ethanol causes triglyceride accumulation in hepatocytes, and might also provide a second hit to livers that have already accumulated fat, via production of reactive oxygen species and initiation of liver inflammation. R
Triglycerides
Bacterial enzymes aid in digestion of otherwise indigestible dietary polysaccharides and extraction of calories from the host's diet.
Enteric bacteria suppress the synthesis of fasting-induced adipocyte factor and secretion from the small intestine, resulting in increased activity of lipoprotein lipase (LPL), and increased accumulation of triglycerides in the liver. R
This process provides a direct link between the intestinal microbiome and fat deposition in the liver.
Choline

Choline is another important metabolite that has been implicated in the pathogenesis of NAFLD and NASH.
High-fat diets lead to formation of intestinal microbiota that convert dietary choline into methylamines, reducing circulating plasma levels of phosphatidylcholine to produce similar effects of choline-deficient diets and causing NASH. R
Phosphatidylcholine is necessary for the assembly and secretion of very-low-density lipoprotein (VLDL). R
Microbiota-induced choline deficiency therefore results in triglyceride accumulation in hepatocytes, secondary to lower hepatic secretion of VLDL, whereas the increase of plasma level of trimethylamine (TMA) and its hepatic metabolism to trimethylamine-N-oxide (TMAO) have been linked to atherosclerosis and cardiovascular disease. R
A single-nucleotide polymorphism in the promoter region of PEMT (rs12325817), which affects de novo synthesis of phosphatidylcholine, is required for development of a fatty liver. R
Endotoxemia
Children with NAFLD had markedly higher serum concentrations of endotoxin than control subjects. R
Genes

No, not jeans...
FAAH
More research to come.
PEMT
Polymorphisms of PEMT can increase the r risk of nonalcoholic fatty liver disease. R
PNPLA3
PNPLA3 (patatin like phospholipase domain containing 3) may be involved in the balance of energy usage/storage in adipocytes. R
PNAPLA3 is associated with risk of non-alcoholic fatty liver disease 1, cirrhosis, and liver cancer. R
rs738409 C>G inresponse to an oral glucose tolrance test can predict NAFLD. R
SIRT1
SIRT enzymes turn off certain genes that are involved in inflammation, fat synthesis and storage, and blood sugar management. R
SIRT1 inhibits pathways involved in aging (IGF-1and mTOR). R R
A reduction in SIRT 1 and SIRT 3 enzymes lead to increased chance of NAFLD, fatty inflitration of muscle, increased fat production, and insulin resistance. R
TLR4
Signaling via toll-like receptor (TLR4), a receptor for lipopolysaccharide (LPS), in hematopoietic-derived cells is required for the development of liver steatosis, but not for the development of obesity in mice. Mice deficient in sensing pathogen-associated molecular patterns (PAMPs) or downstream signaling are resistant to NASH. R
Patients with NAFLD have significantly increased intestinal permeability and alterations in the intestinal tight junctions, compared to healthy individuals. R
Bacterial overgrowth is particularly important in patients with a leaky gut because it increases the luminal amount of PAMPs.
Histamine
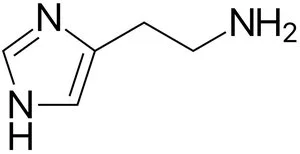
Histamine comes from food and mast cells.
Histamine/H2R signaling might have a protective role in the initial phase during NAFLD progression, correlated with cholesterol and BA metabolism in the liver/intestine. R
Cytokines
TGF-β
TGF-β is a cytokine/growth factor with immunosuppressive, anti-inflammatory, and pro-fibrotic properties. R
In the liver, TGF-β1 is the most abundant isoform, and it is secreted by immune cells, stellate cells, and epithelial cells. R
TGF-β1 plays a pivotal role in hepatic fibrosis by mediating the activation of stellate cells and their production of extracellular matrix proteins. R R R
Kupffer and stellate cells produce TGF-β1, which induces the transformation of resting stellate cells to myofibroblasts. R
In experimental models of hepatic fibrosis induced by CCl4 or schistosomiasis, expression of TGF-β1 is upregulated. R R R
In patients with liver fibrosis, the expression of TGF-β1 mRNA is increased. R R R
The upregulation of TGF-β1 is an early molecular step in the progressive fibrotic steatohepatitis. TGF-β1 levels are increased in patients with NASH as compared to hepatic steatosis. Thus, the measurement of serum levels of TGF-β1 might be useful to distinguish NASH patients in the spectrum of NAFLD. R
Polymorphisms that induce high angiotensinogen and TGF-β1 are associated with advanced hepatic fibrosis in obese patients with NAFLD. R
TNF-α
TNF-α is an inflammatory mediator secreted by several inflammatory cell types, including monocyte/macrophages, neutrophils, and T-cells, but also by many other tissues, such as the endothelium, adipose tissue, or neuronal tissue.
In the liver, TNF-α is secreted directly by hepatocytes and Kupffer cells or indirectly by abdominal fat. R
In Animals
Adipose tissue represents an important source of obesity-induced inflammation, notably by the expression of TNF-α, which can induce inflammation and insulin resistance.
Obese mice lacking TNF-α showed an improved insulin sensibility. R
Mice treated with the anti-TNF-α drug, thalidomide, showed some improvements in the hepatic alterations mediated by a high-fat diet. R
In Humans
TNF-α levels are shown to be higher in obese than in lean individuals, and are correlated with insulin resistance. R R
There is increased TNF-α expression in the liver and adipose tissue in NASH patients with significant fibrosis in comparison with those with a slight or nonexistent fibrosis. R
It is controversial
Some studies did not show any correlation between insulin resistance and TNF-α levels, R R whereas two clinical studies, using an antagonist and an anti-TNF-α antibody, did not show any improvements in insulin sensitivity. R R But in another study, they did not observe any difference in circulating levels of TNF-α between patients with NAFLD as compared to controls without NAFLD. R
Interleukins
IL-6
IL-6 activates several cells, such as immune cells, hepatocytes, hematopoietic stem cells, and osteoclasts. R
IL-6 has a wide range of biological functions, including induction of inflammation and oncogenesis, regulation of immune response, and support of hematopoiesis. R
IL-6 is considered as a hepatoprotector in liver steatosis, capable of reducing oxidative stress and preventing mitochondrial dysfunction. R R It confirmed in other models of liver disease, such as ischemic preconditioning models and in liver regeneration after partial hepatectomy in mice. R R R R
IL-6 is a key element in the acute phase response, mediating the synthesis of several acute phase proteins (such as C-reactive protein and serum amyloid A). R
In diet-induced obese mice, treatment with IL-6 antibodies improved sensitivity to insulin. R
IL-6 is considered as a predictor marker of insulin resistance and cardiovascular diseases. decreased IL-6 concentrations are associated with weight loss and insulin resistance improvement. R
Serum IL-6 levels are higher in animal models and patients with NAFLD. R R R
In humans with NASH, a positive correlation between IL-6 expression in hepatocytes and the severity of NAFLD has been observed. R
IL-6 could improve hepatic regeneration and repair, it could also sensitize the liver to injury, stimulate hepatocyte apoptosis, induce insulin resistance, and participate in NASH development.
IL-6 pathway neutralization with tocilizumab, a specific antibody against the IL-6 receptor, enhanced hepatic steatosis, but improved liver damage in mice with methionine choline deficient (MCD) diet-induced NASH. R
Upregulation of IL-6, but also severe suppression of hepatic IL-6/signal transducer and activator of transcription 3 signaling may lead to the progression of NASH.
IL-10
IL-10 is considered as an anti-inflammatory cytokine that regulates the inflammation in several organs and tissues in physiological or pathological situations. R
It inhibits T cell-, monocyte-, and macrophage-mediated functions.
A study using IL-10-deficient mice fed on high fat diet, suggested that endogenous IL-10 is protective for hepatic steatosis, but not for concomitant insulin resistance. R
The inhibition of IL-10 led to increased expression of pro-inflammatory markers (TNF-α, IL-6, IL-1β, and F4/80) and impaired insulin signal transduction and steatosis. R
The inverse correlation between IL-10 levels and metabolic syndrome in obese woman suggests a potential IL-10-mediated benefit in metabolic syndrome patients also affected by NAFLD. R
Chemokines
Chemokines (chemotactic cytokines) are small heparin-binding proteins known to induce mainly leukocyte trafficking, growth, and activation in inflammatory sites. R R
Chemokines and their receptors have been implicated in multiple inflammatory diseases, such as atherosclerosis, multiple sclerosis, psoriasis, and insulin resistance. R
Expression of several chemokines and chemokine receptors has been shown to be upregulated in the livers of obese patients with severe steatosis and NASH. R
Inflammatory processes are crucial in the potential progression of NAFLD; therefore, chemokines might also play a pivotal role in NAFLD pathophysiology. R
CCL2
CCL2 is a potent chemoattractant that is principally secreted by macrophages and, to a lesser extent, by activated endothelial cells, smooth muscle cells, and hepatic stellate cells. R R R R
Monocyte/macrophage accumulation in the steatotic liver is reduced in mice fed on high-fat diet and who are deficient for CCL2 or CCR2 genes. R
CCL2 deficiency in mice fed on a high-fat diet decreases insulin resistance and hepatic steatosis. Mice overexpressing CCL2 in adipose tissue present increased insulin resistance and hepatic triglyceride levels. R
CCL2 is upregulated in the livers of animals with high-fat diet-induced NASH. R This pathophysiological aspect of CCL2 directly contributed to the lipid accumulation in hepatocytes via the activation of peroxisome proliferator-activated receptor α gene expression. R
Obesity upregulates CCL2 expression in hepatocytes, leading to the recruitment of CCR2-positive myeloid cells and thus, promoting hepatosteatosis. R
Inhibiting the CCL2/CCR2 pathway in several mouse models of metabolic diseases significantly improves obesity, insulin resistance, hepatic steatosis, and inflammation in the adipose tissue. R R R
CCL2 expression is positively correlated with liver fat content in patients with NAFLD. R
CCL5
CCL5 is involved in several chronic immune-inflammatory diseases, such as atherosclerosis, acute myocardial infarction, myocarditis, rheumatoid arthritis, and multiple sclerosis. R R
It is secreted by various cells, such as endothelial cells, smooth muscle cells, macrophages, or hepatic stellate cells. This chemokine is mainly involved in migration of T cells, monocytes, neutrophils, and dendritic cells through binding to its cognate transmembrane receptors, CCR1, 3 and 5.
Hepatic cells are both the target and source of CCL5. R R
Antagonism of CCL5 on receptor CCR5 improved experimental liver fibrosis in mice, indicating that CCL5 is a promising therapeutic target to reduce NAFLD. R
CXCL8/IL-8
CXCL8/IL-8 is a CXC chemokine produced by several cell types, including inflammatory and endothelial cells. R
The major role of this CXCL8 is to orchestrate neutrophil recruitment within inflamed tissues.
Serum levels of CXCL8 are higher in NAFLD patients as compared to obese and non-obese patients. R
CXCL8 serum levels are independently associated with NASH. R
Other CXC Chemokines
CXCR3 is found at high levels on activated T cells, memory T cells, and natural killer cells. R
CXCL9 and CXCL10 mainly induce the migration of these cell types. In the liver, endothelial cells highly express CXCL9 leading to the transmigration of the CXCR3-expressing lymphocytes. R
High levels of CXCL9 are found in the livers of patients with NASH. R
Cannabinoids

The endocannabinoid system (ECS) plays an important role in various liver diseases including viral hepatitis, NAFLD, alcoholic liver disease, hepatic encephalopathy, and autoimmune hepatitis. R

ECs are synthesized on demand, act locally, and are rapidly degraded by specific enzymes. R
ECs are involved in numerous physiological and pathophysiological processes in chronic liver diseases (CLDs).
The upregulation of cannabinoid receptors CB1 and CB2 in NAFLD, alcoholic liver disease, autoimmune and viral hepatitis, I/R, cirrhosis, and related disturbances suggests using CB1 to promote and CB2 to protect from liver damage. R RR

Deletion of CB1 improved hepatic fibrosis and steatosis induced by carbon tetrachloride or high-fat diet, whereas the lack of CB2 increased collagen deposition, liver fat, and enhanced inflammatory scores. R
CB1 blockage by SR141716 (rimonabant) and AM6545, or CB2 agonism with JWH-133 and HU-308 in NAFLD, alcoholic steatohepatitis (ASH), and cholestasis models showed beneficial effects, as reflected by improved hepatic fibrosis, steatosis, inflammation, and liver regeneration, as well as normalization of serum liver enzymes levels, triglycerides, free fatty acids, and cholesterol.
In animal studies:
CB1 antagonists - cirrhosis, high fat diet, biliary fibrosis, alcoholic hepatitis, I/R
Improved fibrosis, steatosis, ascites, hypotension, portal pressure, cardiac contractility, fatty acids oxidation, insulin and leptin sensitivity, I/R-related tissue damage, increased adiponectin
Improved liver damage, reduced obesity and leptin resistance
CB2 agonists - Acute liver injury, alcoholic liver disease, I/R, autoimmune hepatitis, biliary fibrosis, cirrhosis
Suppressed histopathological and apoptotic liver damage, reduced inflammation, oxidative stress, fibrosis and bacterial translocation
With metabolic liver disease/high-fat diet - Induced insulin resistance and steatosis
Suppressed hepatic inflammation and fibrogenesis
In human studies:
CB1 and CB2 agonism - HCV/HIV
Promoted fibrosis progression rate, steatosis severity, glucose intolerance (?), lowered insulin resistance risk
CB1- and CB2-independent, GPR55 antagonist - Hepatic encephalopathy
Improved cognitive and motor function, neuroinflammation
CB1 antagonists - Obesity, metabolic syndrome, NASH, T2D
Reduced the prevalence of metabolic syndrome, body weight, TG, waist circumference, ALT, AST, GGT; increased HDL cholesterol
Cannabinoid Receptors as Targets in CLD

Opposing therapeutic effects of ECS modulation. R
Two non-psychoactive cannabinoids reduce intracellular lipid levels and inhibit hepatosteatosis. R
CB1 as a key mediator of insulin resistance and liver lipogenesis in both animals and humans. On the other hand, CB2 receptors have been shown to promote inflammation with anti-fibrogenic properties. R
In non-alcoholic fatty liver disease, CB2 receptor activation is associated with the development of fatty liver. R
In mouse models of liver fibrosis, activation of CB1 receptors on hepatic stellate cells is fibrogenic, and CB1 blockade slows the progression of fibrosis. R
Click here to read more about CB receptors and the endocannabinoid system.
Drugs and Supplements To Take
Drugs
Treatment with pentoxifylline (a molecule inhibiting TNF-α) decreased the serum levels of aminotransferases and displayed hepatic beneficial effects in patients with NASH. R
Dihydroartemisinin protects the liver in alcoholic liver disease. R
Preclinical studies suggest that OCA (obeticholic acid) improves hepatic steatosis, inflammation and fibrosis. R
LY2405319 treatment attenuated symptoms of steatohepatitis, lowered AST and ALT serum levels, decreased mRNA expression of TGF-β1 and type-I collagen, and decreased phosphorylation of NF-kB p65, JNK1/2, and p38. R
Mice treated with thalidomide showed some improvements in the hepatic alterations mediated by a high-fat diet. R
Telmisartan is beneficial for NAFLD. R
Supplements
Melatonin improved rats with NAFLD (while on a high fat diet). R
Omega 3's are hepatoprotective in NAFLD. R
Vitamin E therapy demonstrated a significant improvement in steatosis, inflammation, ballooning, and resolution of steatohepatitis in adult patients with aggressive NASH. R
NAC (n-acetyl-cysteine) in combination with metformin could be beneficial for the treatment of NAFLD and prevention of NASH. R
If you are an undermethylator, supplement with SAMe. R
Milk Thistle reduced the inflammation in NALFD. R
Phospholipids influences membrane- dependent cellular functions and shows anti-inflammatory, antioxidant, antifibrogenic, anti apoptotic, membrane-protective, and lipid-regulating effects. R
Resveratrol helps, but is very week.R
Treatment
1. Increase Nitric Oxide
TRPV4 which is a part of the body's defense system, is able to activate the release nitric oxide. R
Nitric oxide blocks one of the enzymes (CYP2E1) that is a major contributor to non-alcoholic liver disease and its progression. R
Inhibiting the TRPV4 ion channel can induce hepatoxicity (such as taking acetaminophen or alcohol). R
Click on this line to learn the many ways to increase nitric oxide.
You also want to increase Adropin levels, as it has been shown to be low in NAFLD patients. R
2. Treat Any Underlying Pathogens and SIBO
Fatty liver disease has been linked to intestinal permeability. R
These 3 tests will help you identify underlying pathogenic dysbiosis and SIBO.
3. Treat Inflammation & Insulin Problems w/ Prebiotics
Oligofructose consumption attenuates the inflammation within hepatocytes.R
It also decreases ghrelin concentration.
Ghrelin is an orexigenic hormone which stimulates food ingestion. R
It reduces the risk of insulin resistance, diabetes and obesity development in animals fed a high-fat diet. R
This is what they used this the study. This is very similar and easier to get.
4. Lose Weight
When the patients lost weight, they restored liver fat to normal. R
5. Diet
Simply: cut out alcohol/sugar/MSG, eat high amounts of MUFAs/PUFAs, and fiber, and moderate protein. R
Cut down on sugar (and eliminate it):
Dietary sugars, particularly fructose, can cause an inflamed liver, fibrosis and steatosis. R RR R RR RR
High simple carbohydrate diet the liver quickly accumulates fat. R
Risk of metabolic syndrome is not seen in subjects who do not consume high-fructose. R R
Drinking kefir may prevent the problems with fructose though. R
Eat a MUFA-rich diet:
It inhibitedTNF-α production. R
It increases glucose uptake in fat tissue and reduces visceral fat. R R
Omega 3's (in a good ratio to 6's) decreased steatosis R RRR
MUFA-rich diet (Monounsaturated fatty acids) R
Avocado
Oils - olive R
Walnuts R
(lower amounts of these) nuts and seeds - almonds, cashews, peanuts, macadamia nuts, pecans, pine nuts, pistachios, pumpkin seeds, sesame seeds
PUFA-rich diet (Polyunsaturated fatty acids) R
Sea Fish
Green leafy vegetables
Rapeseed oil
Flax seeds
Protein deficiency, as well as malnutrition, can lead to NASH development. R
Protein intake is essential for the regeneration of hepatocytes and supplies crucial amino acids that prevent excessive fat accumulation within hepatocytes. R
Moderate protein intake (25% of total calories as protein) is as efficient in reducing body fat content as a high-protein diet is.
It is said that a moderate protein intake is optimum for patients with NAFLD. R
MSG R
MSG increases the risk of fatty liver development (enhancing inflammation and dysplasia within hepatocytes.
MSG contributes to the rise in free fatty acids and triglyceride serum concentrations.
MSG causes deterioration in fatty acid β-oxidation, bile acid synthesis and lipid storage.
Diet is a lifestyle. You didn't develop liver problems overnight. Your problems are not going to fix overnight. Got it?
Nutrients
Cobalt, copper, iodine, lithium, manganese, selenium, and zinc has been seen to be reduced in rats with NAFLD. R
More Research
In regards to fructose, Genetically Engineered Escherichia coli Nissle 1917 Synbiotics Reduce Metabolic Effects Induced by Chronic Consumption of Dietary Fructose R
The accumulation/deposition of fat within the liver is essential for diagnosis of NAFLD and might be associated with alterations in the hepatic and systemic inflammatory state. R
Liver disease has long been associated with qualitative and quantitative (overgrowth) dysbiotic changes in the intestinal microbiota. R
Diet- and host-induced changes in the intestinal microbiota contribute to the onset of NAFLD and NASH. Changes in the intestinal microbiota can affect the liver via translocated microbial products or absorbed bacterial metabolites.
Direct host–microbiota interactions in the intestine alter intestinal homeostasis, affecting the liver as distant organ.
Total bacterial counts in the feces, based on real-time PCR, did not differ between healthy subjects and persons with NAFLD or NASH. R
Obesity may maintain a positive feedback loop that promotes Th17 survival in the inflamed liver. R
Immune Imbalances in Non-Alcoholic Fatty Liver Disease: From General Biomarkers and Neutrophils to Interleukin-17 Axis Activation and New Therapeutic Targets. R
The degree of fat content in the liver correlated with the degree of DPP4 gene methylation and the amount of enzymes produced by the liver. R
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Quercetin
500mg 2x/day
Vitamin D3 + K2
5000 IU + 200mcg/day
Magnesium Glycinate
400mg at bedtime