Homocysteine: The Methylation Marker That Links B Vitamins To Your Heart And Brain
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.
Homocysteine is a sulfur amino acid your body makes every time it runs the methylation cycle, and when it climbs it tracks with a damaged endothelium, a faster-shrinking brain, and a stalled one-carbon economy.
In this post, we will discuss what homocysteine actually is, why it rises through the methylation and transsulfuration pathways, what elevated levels do to your heart and brain, how to lower it with the right B vitamins, how to test it, and which genetics keep it high.

What Homocysteine Is
Homocysteine (Hcy) is a sulfur-containing amino acid that is not built into proteins and does not come from food.
It is made entirely inside your cells as a byproduct of methylation, the pathway that donates methyl groups to your DNA, neurotransmitters, hormones, and detox enzymes.
Every methylation reaction starts with S-Adenosylmethionine (SAMe), the universal methyl donor.
When SAMe hands off its methyl group it becomes S-Adenosylhomocysteine (SAH), and SAH is then hydrolyzed to homocysteine.
So homocysteine is the exhaust of the methylation engine, and its level in your blood reflects how well you are recycling that exhaust back into useful fuel.
Your body has two ways to clear it: rebuild it back into methionine (remethylation), or burn it off down a separate disposal route toward glutathione (transsulfuration).
When either route is bottlenecked, homocysteine accumulates in plasma.
Fasting total plasma homocysteine below roughly 7 to 8 micromol/L is a reasonable optimal target, standard lab reference ranges call anything under 15 micromol/L "normal," and a growing body of work argues the real threshold for disease risk sits closer to 10 micromol/L. R
Clinically, hyperhomocysteinemia is graded as mild at 15 to 30 micromol/L, moderate at 30 to 100 micromol/L, and severe above 100 micromol/L, with severe values usually pointing to a genetic enzyme defect. R
For a full walkthrough of the methyl-donor economy that produces homocysteine in the first place, see methylation.
Why It Rises: Methylation And Transsulfuration
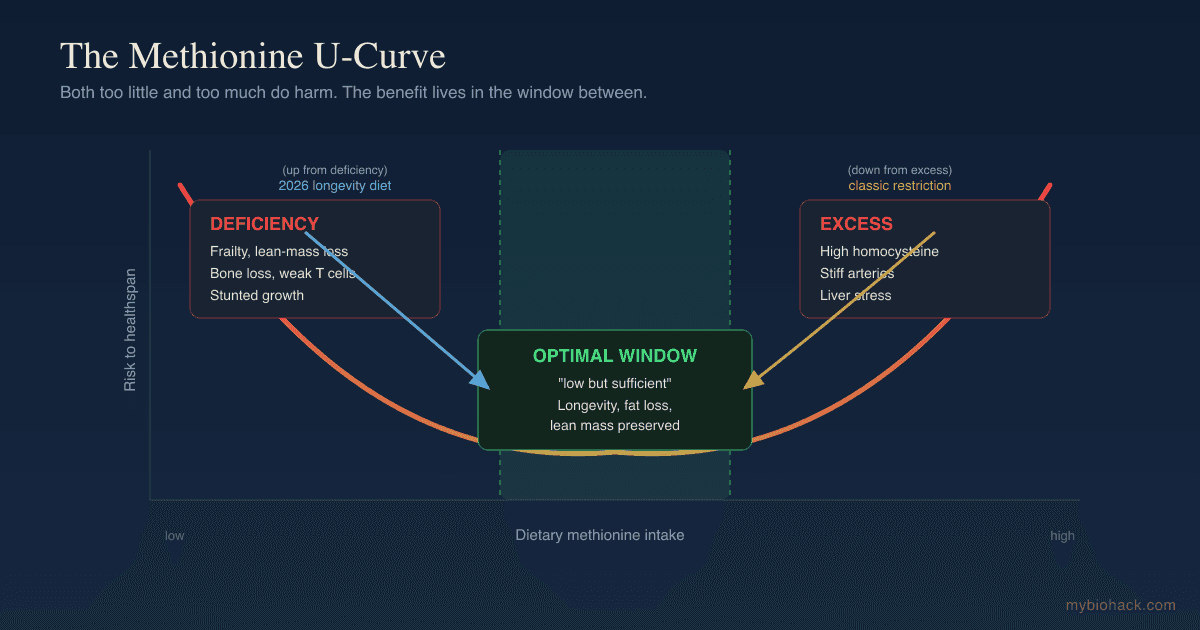
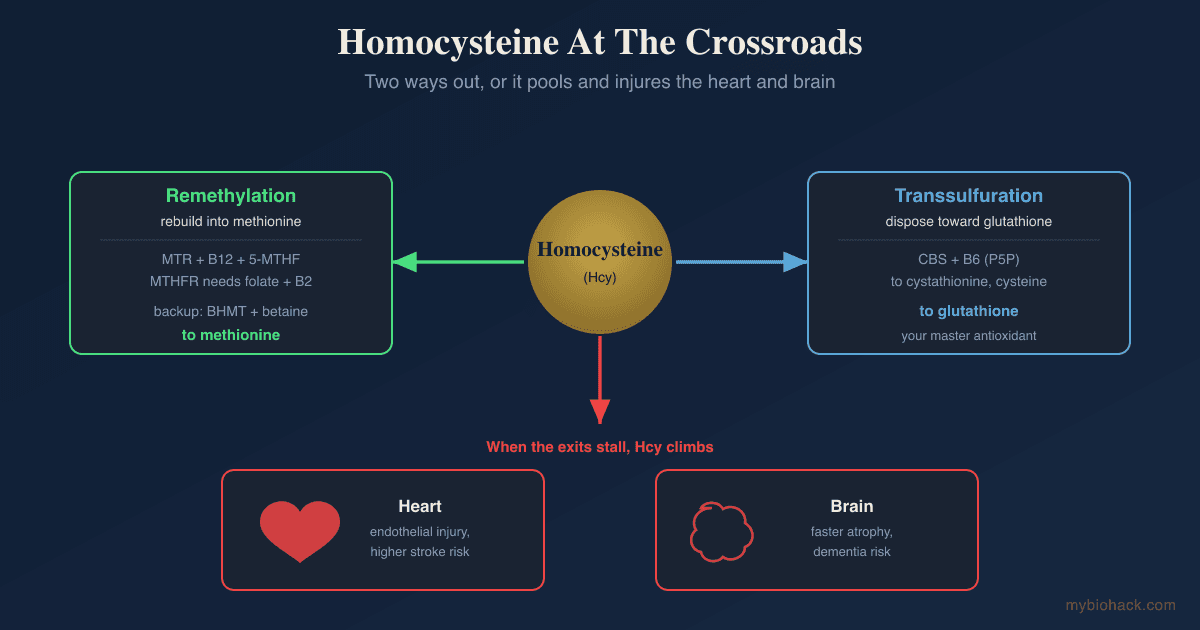
Homocysteine sits at a metabolic crossroads with three exits, and it rises whenever the exits are blocked.
Remethylation (The Folate And B12 Route)
The main exit rebuilds homocysteine into methionine so the methylation cycle can run again.
This step is run by Methionine Synthase (MTR), which requires methylcobalamin (vitamin B12) as its cofactor and pulls a methyl group off 5-Methyltetrahydrofolate (5-MTHF), the active circulating form of folate. R
That 5-MTHF is produced by the enzyme Methylenetetrahydrofolate Reductase (MTHFR), which itself needs riboflavin (vitamin B2) as a cofactor.
So a single remethylation step depends on three separate B vitamins working in series: folate, B12, and B2.
Take out any one of them and homocysteine climbs, which is why folate deficiency and B12 deficiency are the two most common non-genetic causes of elevated homocysteine. R
Remethylation (The Betaine Backup Route)
The liver and kidneys have a second, folate-independent remethylation route.
Betaine-Homocysteine Methyltransferase (BHMT) uses betaine (trimethylglycine) as the methyl donor to convert homocysteine back to methionine.
This is why betaine and its precursor choline both lower homocysteine, and why this backup matters most when the folate route is impaired. R
Transsulfuration (The Disposal Route Toward Glutathione)
The third exit does not recycle homocysteine at all, it commits it to permanent disposal.
Cystathionine Beta-Synthase (CBS) condenses homocysteine with serine to form cystathionine, the first step of the transsulfuration pathway, and this reaction requires Pyridoxal-5-Phosphate (P5P), the active form of vitamin B6. R
Cystathionine is then converted to cysteine, the rate-limiting building block for glutathione, your master antioxidant, and for taurine and hydrogen sulfide. R
This is the elegant part of the biology: the same molecule that damages your vessels when it pools is also the raw material for your most important antioxidant when it flows.
Elevated homocysteine is therefore rarely one broken enzyme, it is usually a combined shortfall of B12, folate, B2, and B6 sitting on top of high methylation demand.
Cardiovascular Effects
Elevated homocysteine is one of the most consistently reported independent risk markers for vascular disease, and here is where the honest nuance begins.
The Observational Signal Is Real
In a meta-analysis of 30 prospective and retrospective studies, a 25 percent lower usual homocysteine level was associated with an 11 percent lower risk of ischemic heart disease and a 19 percent lower risk of stroke. R
Mechanistically this is plausible, because homocysteine damages the endothelium through several convergent routes.
It uncouples endothelial nitric oxide synthase and quenches nitric oxide with reactive oxygen species, which impairs the flow-mediated vasodilation that keeps vessels healthy. R
It inhibits dimethylarginine dimethylaminohydrolase, causing the nitric oxide inhibitor asymmetric dimethylarginine to accumulate, and it drives endoplasmic reticulum stress toward endothelial apoptosis. R
If you want the deeper story on how nitric oxide governs vascular tone, see nitric oxide and how nitric oxide modulates blood pressure.
The Interventional Signal Is Weak For The Heart
Here is the part that most homocysteine content leaves out.
When large randomized trials actually lowered homocysteine with B vitamins, they did not reduce heart attacks.
The HOPE-2 trial gave 5,522 patients with vascular disease folic acid plus B6 and B12 for five years, successfully lowered homocysteine, and found no reduction in major cardiovascular events. R
A Cochrane review of homocysteine-lowering trials reached the same conclusion for myocardial infarction and death, with the one consistent exception being a modest reduction in stroke. R
The clearest stroke signal came from the China Stroke Primary Prevention Trial, where adding 0.8 mg of folic acid to an ACE inhibitor in 20,702 hypertensive adults significantly cut first stroke, with the largest benefit in people with low baseline folate and the MTHFR variant. R
So the honest read for the heart is this: homocysteine is a strong marker of vascular risk, lowering it clearly helps prevent stroke, but there is a big MAYBE on whether it prevents coronary events, and the genetics (below) suggest it may be more of a passenger than a driver for coronary artery disease.
Brain And Dementia
The brain is where the homocysteine story gets much more convincing than it is for the heart.
Homocysteine Predicts Dementia
In the Framingham Study, an elevated plasma homocysteine level was a strong, independent predictor of later dementia and Alzheimer's disease, even after adjusting for age, ApoE genotype, and B vitamin levels. R
For readers carrying the highest-risk dementia genotype, see ApoE4, which stacks with high homocysteine.
B Vitamins Slow Brain Atrophy
The landmark VITACOG trial randomized older adults with mild cognitive impairment to high-dose B vitamins (folic acid, B6, and B12) or placebo for two years and tracked brain volume by serial MRI.
The B vitamins lowered homocysteine and slowed the rate of whole-brain atrophy, with the greatest protection in those who started with the highest homocysteine. R
The Omega-3 Catch
This is the nuance that makes the brain data actionable rather than just interesting.
A follow-up analysis of VITACOG found that the B vitamins only slowed atrophy in participants who also had good omega-3 status.
People with low plasma EPA and DHA got essentially no benefit from the B vitamins, while those with high omega-3 saw brain shrinkage slowed by around 70 percent. R
In other words, lowering homocysteine to protect the brain appears to require adequate omega-3 fatty acids to work, which is a genuine interaction and not a footnote.

How To Lower It
The goal is to feed every enzyme in the homocysteine crossroads and reduce methylation demand, not to megadose a single B vitamin.

1. Methylfolate (The Folate Route)
Folate is the single biggest lever on homocysteine because it fuels the main remethylation step.
Use the active form rather than synthetic folic acid, because folic acid must be reduced by MTHFR before it can be used, and in people with MTHFR variants or high intakes this leaves unmetabolized folic acid circulating in the blood. R
Methylfolate (L-5-MTHF) is already in the active form and bypasses the MTHFR bottleneck entirely.
2. Methylcobalamin (The B12 Route)
Methionine synthase cannot run without B12, so B12 deficiency raises homocysteine even when folate is adequate. R
Methyl-B12 (methylcobalamin) is the coenzyme form used directly by methionine synthase.
Sublingual dosing is useful for anyone with low stomach acid, gut inflammation, or malabsorption.
3. Riboflavin (The Overlooked Cofactor)
MTHFR uses riboflavin (B2) as its cofactor, and this matters enormously for people with the MTHFR 677TT genotype.
In a randomized trial, riboflavin specifically lowered homocysteine in 677TT individuals whose enzyme is riboflavin-starved, an effect invisible in people without the variant. R
This is the most underused homocysteine intervention, and it is nearly free.
4. P5P (The Transsulfuration Route)
If homocysteine is disposed of rather than recycled, it goes down the CBS route, which is B6-dependent. R
P5P (pyridoxal-5-phosphate) is the active form of B6 and supports transsulfuration into glutathione.
5. TMG / Betaine (The Backup Route)
Betaine feeds the BHMT enzyme, the folate-independent way to remethylate homocysteine.
In healthy adults, TMG (trimethylglycine, betaine) lowers fasting homocysteine dose-dependently, by roughly 12 to 20 percent across 1.5 to 6 grams per day. R
A meta-analysis confirmed that at least 4 grams per day reliably reduces plasma homocysteine. R
6. Omega-3 (The Brain Multiplier)
If your reason for lowering homocysteine is the brain, omega-3 status is not optional.
The VITACOG data show B vitamins only slowed brain atrophy in people with adequate EPA and DHA. R
Omega-3 (EPA and DHA) should be considered part of any homocysteine-for-cognition protocol.
7. Reduce Methylation Demand With Creatine
Creatine synthesis consumes roughly half of all methyl groups your body spends, making it the single largest drain on methylation and a major generator of homocysteine. R
Supplementing creatine can spare that methyl demand, and in animal models it lowered homocysteine by around 25 percent, although human data on homocysteine specifically are mixed (there is a big MAYBE here for people, even though the mechanism is real). R
For the broader case on creatine and the brain, see creatine.
What Raises It (Limit These)
- Coffee in large amounts (chronic high intake nudges homocysteine up)
- Folic acid megadoses (leaves unmetabolized folic acid, especially with MTHFR variants) R
- High methionine intake without adequate B vitamins (drives the cycle without clearing the exhaust)
- Hypothyroidism, renal impairment, and certain drugs (methotrexate, metformin, antiepileptics)
Testing
Homocysteine is one of the highest-value, lowest-cost markers you can run, and it is worth pairing with the B vitamins that drive it.
Blood And Urine Markers
Fasting total plasma homocysteine is the core test, and it must be drawn fasting because a recent protein meal transiently raises it. R
I use the Homocysteine test (Quest via Fullscript) as a standalone marker, targeting a fasting value under about 7 to 8 micromol/L rather than merely under the lab ceiling of 15. R
Because homocysteine only tells you it is elevated, not why, the more useful order is the Homocysteine + B12 + Folate panel (Quest via Fullscript), which shows whether the remethylation cofactors are the bottleneck.
For B6 status, which governs the transsulfuration exit, add the Vitamin B6 test (Quest via Fullscript), and for B12 alone the Vitamin B12 test (Quest via Fullscript).
Elevated homocysteine alongside elevated methylmalonic acid points specifically to B12 deficiency rather than folate deficiency. R
Functional Lab Panels
I use the Methylation Profile (Doctor's Data) to see homocysteine in context with methionine, SAMe, and SAH, which reveals whether the whole methylation cycle is turning over or backing up.
For B vitamins and minerals in one comprehensive order, the Nutrient Zoomer (Vibrant Wellness) covers B6, B12, folate, and the cofactors, and the Cellular Zoomer (Vibrant Wellness) reports organic-acid methylation markers.
If cardiovascular risk is the reason for testing, the Cardio Zoomer (Vibrant Wellness) pairs homocysteine's endothelial story with ApoB and lipoprotein markers, which you can read alongside ApoB and lipoprotein(a).
Genetics
Because the enzymes above are genetically variable, the MTHFR test (Quest via Fullscript) checks the two common variants directly, and the broader Methylation Genetics panel (Vibrant Wellness) covers MTHFR, MTR, MTRR, BHMT, and COMT together.
For anyone building a full workup, the general wellness bundle packages the nutrient and cellular panels at a lower combined cost.
Mechanisms Of Action
Simple:
- Homocysteine is the leftover waste from the reaction your cells use to add methyl tags to DNA, brain chemicals, and hormones.
- Your body clears it either by rebuilding it into methionine (needs folate, B12, and B2) or by burning it into glutathione (needs B6).
- When those B vitamins run short, the waste piles up in your blood and irritates the lining of your blood vessels.
- Lowering it clearly helps prevent stroke and helps protect the aging brain when omega-3 is also adequate, but it has not clearly prevented heart attacks in trials.
Advanced:
- The SAMe / SAH methylation cycle generates homocysteine stoichiometrically: every methyl group transferred from SAMe produces one molecule of SAH, which SAH hydrolase converts to homocysteine plus adenosine, so total methylation flux sets the homocysteine load. R
- Remethylation via MTR regenerates methionine using methylcobalamin and 5-MTHF as the methyl source, and impaired MTR reactivation (or B12 deficiency) traps folate as 5-MTHF in the "methyl-folate trap," simultaneously raising homocysteine and functionally depleting usable folate. R
- Transsulfuration via CBS irreversibly commits homocysteine to cysteine and glutathione synthesis through two consecutive P5P-dependent reactions, and low B6 status shifts the balance away from disposal, raising steady-state homocysteine. R
- Endothelial injury proceeds through eNOS uncoupling, ROS-mediated quenching of nitric oxide to peroxynitrite, DDAH inhibition with ADMA accumulation, and ER-stress-driven apoptosis, collectively reducing nitric-oxide-dependent vasodilation. R
- Guanidinoacetate methylation by GAMT for creatine synthesis is the dominant methyl sink, so high creatine-synthesis demand raises homocysteine while dietary creatine can suppress it by reducing GAA available for methylation. R
Genetics
MTHFR: Highest Population Risk
MTHFR encodes methylenetetrahydrofolate reductase, the enzyme that produces the active 5-MTHF used to remethylate homocysteine.
Loss-of-function variants reduce enzyme activity and raise plasma homocysteine, and the enzyme becomes thermolabile and riboflavin-dependent.
rs1801133 (C677T): the TT genotype cuts MTHFR activity by roughly 70 percent, is the classic driver of elevated homocysteine, and was the original "candidate genetic risk factor for vascular disease." R
rs1801131 (A1298C): a milder activity reduction that adds to risk mainly when inherited alongside a 677T allele (compound heterozygosity).
Note the honest genetic counterpoint: a large meta-analysis of MTHFR case-control data found the 677TT genotype conferred little or no excess coronary heart disease risk, implying lifelong moderate homocysteine elevation is more marker than cause for the coronary arteries. R
MTR
MTR encodes methionine synthase, the B12-dependent enzyme that remethylates homocysteine to methionine.
Variants can alter enzyme kinetics and modestly shift homocysteine and coronary risk.
rs1805087 (A2756G): associated with altered plasma homocysteine and studied in relation to coronary artery disease. R
MTRR
MTRR encodes methionine synthase reductase, which reactivates the B12 cofactor on MTR after it becomes oxidized.
A sluggish MTRR variant leaves methionine synthase stuck in its inactive state, raising homocysteine especially when B12 is marginal.
rs1801394 (A66G): reduces reactivation efficiency and interacts strongly with B12 status. R
CBS
CBS encodes cystathionine beta-synthase, the B6-dependent gateway to the transsulfuration (disposal) pathway.
Complete loss of function causes classic homocystinuria with severely elevated homocysteine, while milder variants shift how much homocysteine is disposed of versus recycled.
Severe hyperhomocysteinemia above 100 micromol/L almost always signals a CBS or comparable genetic defect. R
BHMT
BHMT encodes betaine-homocysteine methyltransferase, the folate-independent remethylation route that uses betaine as the methyl donor.
Variants here make the betaine backup less efficient, which is part of why betaine and choline supplementation help some people more than others. R
More Research
- Betaine and choline are interchangeable upstream, because choline is oxidized to betaine, and supplementing phosphatidylcholine lowers both fasting and post-methionine-load homocysteine through the BHMT route. R
- Folic acid fortification of the food supply lowered population homocysteine, but it also raised circulating unmetabolized folic acid, which is one argument for using methylfolate instead of high-dose folic acid in supplements. R
- Homocysteine damages the endothelial glycocalyx layer that protects the vessel wall, which connects it to the broader vascular-injury model behind post-viral and chronic vascular illness, covered in the endothelial glycocalyx and spike protein and the glycocalyx. R
- For biomarker testing I use the Homocysteine + B12 + Folate panel as the first-line workup, then the Methylation Profile when the whole cycle needs to be visualized.
- Riboflavin's homocysteine-lowering effect is genotype-specific and among the most reproducible gene-nutrient interactions in nutrition, yet it is almost never prescribed, which makes B2 status worth checking before assuming folate or B12 is the problem. R
- The interventional paradox remains unresolved: homocysteine is a robust risk marker and lowering it prevents stroke, but the failure of B vitamins to prevent coronary events, combined with the null MTHFR genetic signal for coronary disease, suggests homocysteine may index vascular and methylation health more than it single-handedly causes coronary artery disease. R R
- The brain evidence is stronger than the heart evidence, and it is conditional: B vitamins slowed brain atrophy in high-homocysteine individuals only when omega-3 status was adequate, so the two interventions appear to require each other. R R
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Electrolyte Complex
1 scoop/day
CoQ10
200mg/day
Magnesium Glycinate
400mg at bedtime