Low Dose Naltrexone, TLR4, And The Endorphin Rebound: Three Mechanisms, One Drug, Very Different Doses
By Jacob Gordon, INHC, FMT-CNaltrexone is an opioid antagonist approved at 50 mg per day for alcohol and opioid use disorder. At that standard dose, it blocks opioid receptors continuously and prevents the euphoric effect of opioids and alcohol.
Below 5 mg per day, the pharmacology changes completely. Below 1 microgram per day, it changes again.
This post covers three distinct dose-dependent mechanisms, why they do not overlap, what the evidence actually shows, and where this fits in chronic illness and pain management.

Standard Naltrexone vs Low Dose: The Dose-Dependent Paradox
At the standard 50 mg dose, naltrexone occupies mu-opioid receptors (MOR) for approximately 24 hours, preventing endogenous and exogenous opioids from binding. Continuous opioid receptor blockade at this level suppresses the endogenous opioid system and is used to remove the reward from alcohol and opioids in addiction treatment.
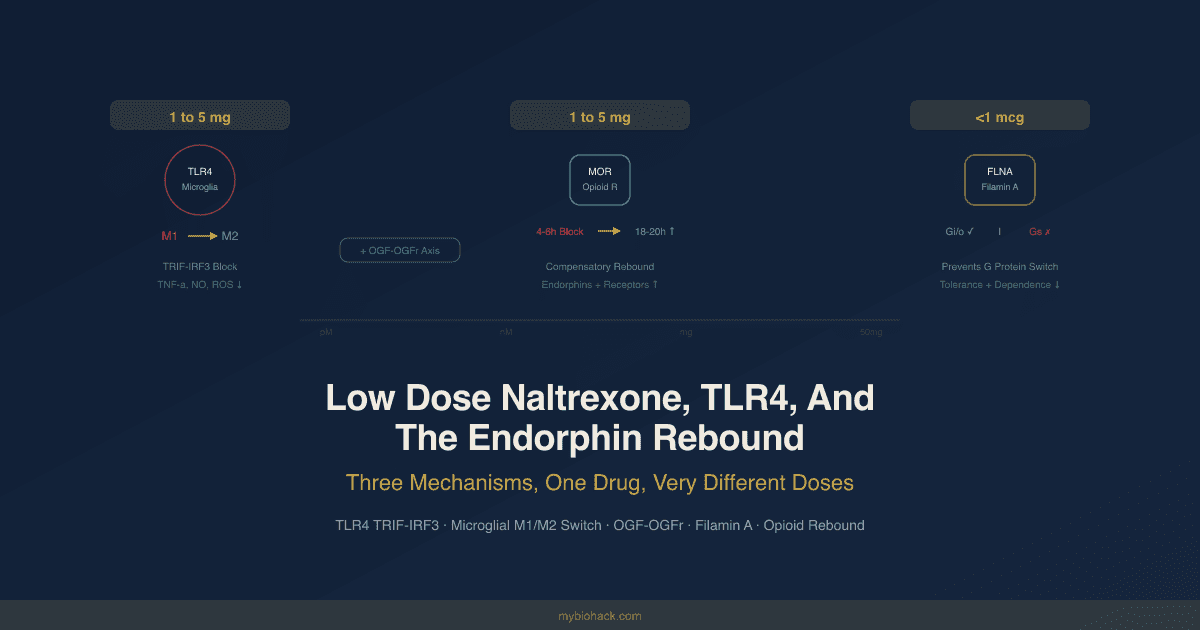
At 1 to 5 mg per day (low dose naltrexone, or LDN), the pharmacology is different in two key ways.
First, at these doses, naltrexone's affinity for the classical mu-opioid receptor drops substantially, while its affinity for Toll-like receptor 4 (TLR4) on microglial cells becomes pharmacologically relevant. This is not a competitive relationship at standard doses; TLR4 binding is negligible at 50 mg. At 1 to 5 mg, TLR4 antagonism becomes the dominant mechanism. R
Second, the brief and partial opioid receptor blockade produced by low doses (lasting approximately 4 to 6 hours before the drug clears) creates a compensatory rebound during which the body upregulates endogenous opioid production and opioid receptor density. This upregulation operates during the remaining 18 to 20 hours when the drug has cleared and opioid receptors are unoccupied. R
At below 1 microgram per day (ultra low dose naltrexone, or ULDN), neither the TLR4 mechanism nor the opioid receptor mechanism is operative. Instead, ULDN binds to a high-affinity site on the scaffolding protein filamin A (FLNA), which modulates how the mu-opioid receptor couples to G proteins, amplifying endogenous and exogenous opioid analgesia and reducing tolerance and dependence. R
These are three distinct mechanisms operating at three non-overlapping dose windows. Understanding which mechanism you are targeting determines the dose, the timing, and the expected clinical effect.
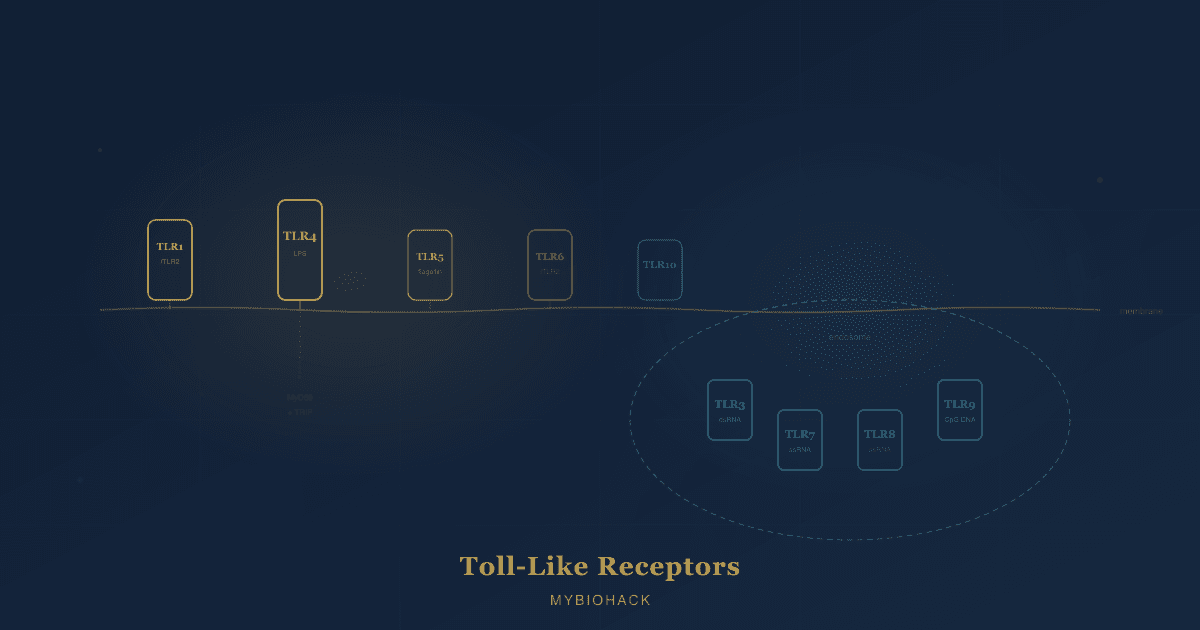
Mechanism 1: TLR4 Antagonism And Microglial Modulation
Toll-like receptor 4 (TLR4) is a pattern recognition receptor that functions as a sentinel of innate immunity. In the periphery, TLR4 on macrophages detects lipopolysaccharide (LPS) from gram-negative bacterial cell walls, triggering an inflammatory response to clear infection. In the central nervous system (CNS), TLR4 is predominantly expressed on microglia, the brain's resident immune cells, which comprise approximately 10 to 15% of all CNS cells. R
Microglia in their resting (M2 or homeostatic) state perform surveillance, synaptic pruning, and tissue maintenance. When TLR4 is activated by bacterial products, damaged tissue signals (damage-associated molecular patterns, or DAMPs), or other inflammatory triggers, microglia shift toward an activated (M1) state and release pro-inflammatory cytokines including:
- Interleukin-1 beta (IL-1beta)
- Interleukin-6 (IL-6)
- Tumor necrosis factor alpha (TNF-alpha)
- Interferon beta (IFN-beta)
- Nitric oxide (NO)
- Reactive oxygen species (ROS) R
This microglial activation is appropriate and time-limited in acute infection. In chronic pain, autoimmune disease, and neurodegeneration, microglial TLR4 signaling can become pathologically sustained, producing a state of chronic neuroinflammation that amplifies pain signals, drives fatigue, disrupts cognition, and accelerates neuronal damage.
TLR4 is not constitutively expressed at high levels in healthy microglia. It is upregulated during inflammation, which creates a feed-forward cycle: inflammation triggers TLR4 upregulation, TLR4 activation drives more inflammation. R
Naltrexone at LDN doses binds TLR4 and blocks this signaling.
The consequence is a shift in the M1/M2 microglial balance back toward the M2 (anti-inflammatory, tissue-supportive) phenotype, reduced pro-inflammatory cytokine output, and attenuation of the neuroinflammatory state that is driving symptoms in a range of chronic conditions. R
The TLR4 Signaling Cascade: MyD88 And TRIF
TLR4 signals through two distinct intracellular pathways downstream of receptor activation.
The MyD88 pathway activates nuclear factor kappa-B (NF-kB) and the MAP kinase cascade (p38, JNK), producing the early inflammatory cytokines IL-1beta, IL-6, and TNF-alpha. R
The TRIF pathway (TIR-domain-containing adapter-inducing interferon-beta) activates interferon regulatory factor 3 (IRF3), producing IFN-beta, NO, and delayed TNF-alpha. R
Naltrexone and naloxone act as TRIF-IRF3 biased TLR4 antagonists. They specifically inhibit the TRIF-IRF3 arm of TLR4 signaling, blocking downstream production of NO, TNF-alpha, and ROS. They do not significantly inhibit TLR4 signaling through NF-kB, p38, or JNK in the same cell models. R
This selective pathway bias has several implications. It means LDN does not broadly suppress innate immune function (which would be dangerous), but rather targets the chronic inflammatory signaling arm that produces sustained tissue damage. The TRIF-IRF3 arm is particularly implicated in neuropathic pain, neurodegeneration, and the pathological pro-inflammatory state of chronically activated microglia. R
In BV-2 microglial cells stimulated with LPS and interferon-gamma (IFN-gamma), naltrexone treatment produced immunometabolic reprogramming, shifting activated microglia away from the oxidative phosphorylation and glycolytic profile of M1 activation and toward a more metabolically quiescent M2 state. This shift was associated with reduced pro-inflammatory cytokine production and reduced TLR4-mTOR (mechanistic target of rapamycin) signaling, since mTOR activity is tied to TLR4-driven microglial activation. R
Stereoselective Proof: (+)-Naltrexone
The most compelling mechanistic evidence that LDN's anti-inflammatory effects are TLR4-mediated and not opioid receptor-mediated comes from stereochemistry.
Classical opioid receptors (mu, delta, kappa) are stereoselective: they recognize only the (-) enantiomer (left-handed mirror image) of opioid molecules. TLR4 is not stereoselective. R
(+)-Naltrexone is the mirror image (dextrorotatory enantiomer) of standard (-)-naltrexone. It has essentially no affinity for classical opioid receptors and cannot produce opioid antagonism. But (+)-naltrexone retains full TLR4 antagonist activity, blocking TRIF-IRF3 signaling, reducing NO, TNF-alpha, and ROS production in microglia, and reversing neuropathic pain in animal models. R
This is the critical experiment that separates the two mechanisms. Because (+)-naltrexone cannot interact with opioid receptors but still produces anti-inflammatory and analgesic effects, those effects are definitively attributable to TLR4 antagonism rather than to any opioid system activity. R
It also means that theoretically, (+)-naltrexone could deliver the anti-inflammatory benefits of LDN without any effect on the endogenous opioid system, allowing cleaner mechanistic separation and use in patients on opioid analgesics without disrupting their pain medication. R
Drug development efforts are now producing (+)-naltrexone-inspired TLR4 antagonists with nanomolar potency, far exceeding the TLR4 affinity of naltrexone itself. The compound CIAC101, an isobutyl-substituted (+)-naltrexone derivative, showed approximately 6,200-fold higher TLR4 antagonism than (+)-naltrexone, dose-dependently blocking microglial activation, reducing NF-kB and pro-inflammatory gene expression, and attenuating methamphetamine behavioral sensitization in animal models at 0.2 mg/kg. R
Mechanism 2: The Opioid Rebound And Endorphin Upregulation
The second mechanism of LDN operates on the classical opioid receptor system and produces effects through a rebound homeostatic response.
When naltrexone is taken at a low dose in the evening (typically), it briefly blocks opioid receptors for approximately 4 to 6 hours. The brain interprets this as a sudden deficit in opioid signaling. The homeostatic compensatory response is to increase production of endogenous opioids (primarily beta-endorphin and met-enkephalin) and to upregulate opioid receptor density and sensitivity. R
After the naltrexone clears (approximately 4 to 6 hours after administration), the upregulated opioids now interact with upregulated receptors during the remaining 18 to 20 hours of the day. The net effect is enhanced endogenous opioid tone throughout most of the day, not just during the period of drug activity. R
What enhanced endorphin tone does:
Endogenous opioids are not just analgesics. They regulate immune function, mood, autonomic nervous system tone, and cellular proliferation. Beta-endorphin in particular has well-documented immunomodulatory effects, influencing natural killer (NK) cell activity, T cell proliferation, and the release of pro-inflammatory cytokines from immune cells. R
In patients with multiple sclerosis (MS), serum enkephalin (met-enkephalin, or opioid growth factor) levels are reduced compared with subjects with other neurological disorders. LDN treatment restored serum enkephalin levels in MS patients and correlated with improved behavioral scores in experimental autoimmune encephalomyelitis (EAE) models, an animal model of MS. R
The practical implication: LDN upregulates the body's own pain and mood management system by exploiting the homeostatic over-response to brief receptor blockade, producing a day-long amplification of endogenous opioid activity from a dose that is pharmacologically active for only a fraction of the day. R
Mechanism 3: The Opioid Growth Factor Axis
This is the least discussed of LDN's three mechanisms but has the most research behind it from a specific research group and is particularly relevant for cancer biology and autoimmunity.
Opioid growth factor (OGF) is not a classical analgesic opioid. It is the endogenous pentapeptide [Met5]-enkephalin (methionine enkephalin), and its primary biological role is as a constitutively active inhibitory growth regulator. R
OGF binds the OGF receptor (OGFr), also known as the zeta opioid receptor, which is structurally and pharmacologically distinct from the classical mu, delta, and kappa opioid receptors. OGFr is located on the outer nuclear envelope (not the cell surface), and the OGF-OGFr complex is internalized via clathrin-mediated endocytosis and transported to the nucleus where it exerts its effects. R
What OGF-OGFr signaling does:
The OGF-OGFr complex upregulates cyclin-dependent kinase inhibitors (CDKIs) p16 and p21, which arrest cells in the G1/S phase of the cell cycle. This slows cellular proliferation in a dose-dependent, receptor-mediated, reversible manner that is not apoptosis or necrosis. It is a growth brake, not a kill signal. R
The LDN connection:
Naltrexone at low doses transiently blocks OGFr as well as classical opioid receptors. This brief blockade interrupts the tonic OGF-OGFr inhibitory signal. The homeostatic response is to upregulate both OGF production and OGFr receptor expression. After the naltrexone clears, the upregulated OGF interacts with upregulated OGFr during the 18 to 20 hours of no blockade, producing enhanced inhibitory growth regulation. R
Implications for cancer:
In multiple cancer cell lines and animal tumor models, OGF-OGFr signaling slows tumor growth, reduces angiogenesis, and inhibits metastatic potential. OGF was studied in ovarian cancer, head and neck squamous cell carcinoma (HNSCC), pancreatic cancer, and triple-negative breast cancer. Administration of LDN or direct OGF peptide treatment delayed tumor growth, extended time to measurable tumor, and in some models produced additive or synergistic effects with cisplatin. R R
Implications for autoimmunity:
Both T and B lymphocytes express OGFr and respond to OGF by reducing proliferation through the p16/p21 pathway. In T lymphocyte cultures stimulated with phytohemagglutinin (PHA) to model immune activation, OGF significantly decreased DNA synthesis without inducing apoptosis or necrosis, suggesting OGF acts as an immunosuppressant specifically by slowing lymphocyte proliferation rather than killing cells. R
LDN, by upregulating the OGF-OGFr axis, may dampen pathological lymphocyte hyperactivation in autoimmune conditions without the broad immunosuppression associated with corticosteroids or disease-modifying antirheumatic drugs (DMARDs). R
Ultra Low Dose Naltrexone: Filamin A And The G Protein Switch
Ultra low dose naltrexone (ULDN) operates below 1 microgram per day (typically in the picogram to nanogram range). At these concentrations, neither TLR4 nor classical opioid receptors are meaningfully engaged. The operative target is filamin A (FLNA).
Filamin A background:
FLNA is a 300 kDa scaffolding protein that cross-links cytoplasmic actin filaments and, more importantly for ULDN pharmacology, physically interacts with the mu-opioid receptor (MOR) at the intracellular C-terminal domain. R
The MOR-FLNA interaction normally functions as a molecular clutch that holds MOR in a Gi/o-coupled signaling mode. Gi/o coupling produces the classical opioid effects: analgesia, sedation, euphoria, and inhibition of adenylyl cyclase.
What chronic opioid exposure does:
After acute or chronic opioid stimulation, MOR undergoes a conformational change that allows it to switch its G protein coupling from Gi/o to Gs. Gs coupling to MOR produces excitatory signaling via adenylyl cyclase and cAMP upregulation, which drives:
- Opioid tolerance (reduced analgesic efficacy)
- Hyperalgesia (increased pain sensitivity)
- Opioid dependence
- Prolonged action potentials in pain-signaling neurons R
This Gi/o to Gs switch is the molecular basis for opioid tolerance.
How ULDN prevents it:
FLNA has a high-affinity binding site for naloxone and naltrexone at a specific pentapeptide sequence in the C-terminal domain (FLNA residues 2561 to 2565), with affinity in the picomolar range (approximately 4 pM). R
When ULDN binds this site, it prevents the critical MOR-FLNA interaction that would otherwise allow Gs coupling to occur. MOR remains locked in its Gi/o-coupled mode. Analgesic efficacy is maintained and enhanced; tolerance, hyperalgesia, and dependence are prevented or reduced. R
The critical dose window:
FLNA also contains a low-affinity binding site for naltrexone at approximately 834 pM, about 200 times lower affinity than the high-affinity site. If both sites are occupied (which happens at higher naltrexone doses), the benefit of the high-affinity site binding is abolished. This explains the extremely narrow dose window for ULDN: slightly above the picomolar-effective dose, the low-affinity site begins to be occupied and the beneficial effect disappears before the LDN mechanisms start to operate. R
This non-monotonic dose-response relationship, where a very low dose works but a slightly higher dose does not, and then a much higher dose (LDN range) produces different effects through entirely different mechanisms, is unusual in pharmacology and explains why ULDN has been difficult to study and often dismissed.
Practical applications of ULDN:
- Postoperative pain management: ULDN co-administered with morphine or other opioids reduces total opioid requirement and incidence of opioid-related side effects including sedation, nausea, and respiratory depression
- Opioid-sparing: in patients requiring chronic opioid analgesics, ULDN maintains analgesic efficacy at lower doses
- Tolerance prevention: ULDN co-administered from the start of opioid therapy may prevent the development of tolerance, reducing dose escalation over time R
The commercial product Oxytrex combined oxycodone with 2 micrograms naltrexone per day. In clinical trials, the 2 microgram naltrexone combination (approximately the ULDN range) provided the greatest reduction in pain intensity in both males and females while substantially reducing opioid side effects and dependence potential. R
Clinical Evidence: What Holds Up
The honest summary of the LDN clinical literature is: promising signal, small samples, limited replication, and a growing but still incomplete evidence base.
What we have:
Most LDN clinical studies are small (fewer than 50 participants), unblinded or single-blind, and have not been adequately replicated by independent groups. The majority of benefit is captured in subjective outcomes (self-reported pain, quality of life, fatigue) rather than objective biomarkers.
What the meta-analyses show:
A 2025 meta-analysis of RCTs (randomized controlled trials) in fibromyalgia found LDN superior to placebo for pain in primary analysis (standardized mean difference, or SMD, of -0.61; 95% confidence interval -1.14 to -0.08) and in sensitivity analysis (SMD -0.87; 95% CI -1.28 to -0.46). However, LDN was not superior to placebo for raising mechanical pain threshold. R
A 2024 meta-analysis of eight studies in fibromyalgia found within-group effects were strong (SMD for pain -1.03; SMD for fibromyalgia severity -1.02), but the between-group difference (LDN vs placebo) did not reach significance (SMD -0.50; 95% CI -1.19 to 0.19), with high heterogeneity across trials (I2 of 91 to 95%). R
This means: the data show LDN produces real-world improvements in fibromyalgia patients, but the current trial size and design cannot definitively separate this from placebo effects. More rigorous trials are needed, and several are underway.
Crohn's disease has the strongest objective evidence:
In Crohn's disease studies, LDN has reduced not only self-reported pain but also objective markers including disease activity scores, endoscopic severity scores, and inflammatory biomarkers. Response rates exceeding 80% have been reported in small Crohn's studies, higher than in fibromyalgia. R
The safety profile is genuinely favorable:
Across studies, tolerability of LDN is rated comparable to placebo. Reported side effects are mild and include vivid dreams (most common, related to altered opioid activity during sleep), headache, and occasional nausea. Side effects typically resolve after the first week of use. No serious adverse events have been attributed to LDN in clinical trials. R
The regulatory situation:
LDN is entirely off-label. It requires a compounding pharmacy because no pharmaceutical company has sponsored the large trials needed for approval. The lack of patentability for naltrexone (an old generic drug) is the primary barrier to industry-funded trials, not safety concerns.
Conditions With The Strongest Signal
Fibromyalgia: Consistent subjective improvements in pain, fatigue, sleep, and mood in small trials. Mechanistic coherence: fibromyalgia is characterized by central sensitization driven by glial cell activation and neuroinflammation, the exact targets of LDN's TLR4 mechanism. A 2024 Lancet Rheumatology RCT (naltrexone 6 mg in women with fibromyalgia) reported significant improvements vs placebo. Cytokine panels in LDN-treated fibromyalgia patients show reduced pro-inflammatory cytokines at 8 weeks. R R
Crohn's disease: The best objective evidence for LDN. Studies in both adults and children with moderate to severe Crohn's showed safety, tolerability, and reduced disease activity scores. The anti-inflammatory TLR4 mechanism maps directly onto Crohn's pathophysiology, where dysregulated innate immune signaling through TLR4 on intestinal macrophages and dendritic cells drives mucosal inflammation. The OGF axis upregulation may also reduce pathological mucosal cell hyperproliferation. R
Multiple sclerosis: Reduced fatigue, improved quality of life, and spasticity reduction in several small trials. Reduced serum enkephalin levels in MS patients are restored by LDN treatment. LDN and OGF reduce CNS infiltration by CD4+ T lymphocytes in EAE animal models. R
Complex regional pain syndrome (CRPS): Post-mortem immunohistochemical analysis of spinal cord from a CRPS patient demonstrated upregulation of microglial TLR4 receptors and overactivation of microglia in the dorsal horn at the original injury level. This pathology is mechanistically aligned with LDN's TLR4 microglial modulation mechanism. Case reports show significant symptom improvement. R
Long COVID: An interventional pre-post study found LDN safe and showed improvements in fatigue, cognitive symptoms, and sleep in a long COVID cohort. The neuroinflammatory hypothesis of long COVID (driven by persistent microglial activation and TLR4 signaling from SARS-CoV-2 components) provides mechanistic rationale. R
Cancer (preclinical, early clinical): The OGF-OGFr mechanism has been studied in ovarian cancer, HNSCC, pancreatic cancer, and triple-negative breast cancer in animal models. LDN slowed tumor growth, delayed time to measurable tumors, and showed additive effects with standard chemotherapy in some models. No large human cancer trials have been completed, but the mechanistic rationale is evidence-based and the safety profile permits investigation. R
Practical Dosing And Timing
LDN dosing range: 1 to 5 mg per day (most commonly 1.5 to 4.5 mg)
Titration protocol: Start at 0.5 mg per day for 1 to 2 weeks to assess tolerability. Increase to 1.5 mg for 2 weeks. Increase to 3 mg for 2 to 4 weeks. Target dose for most conditions is 4.5 mg per day. Some patients respond better at lower doses (1.5 to 3 mg); individual dose-finding is appropriate.
Timing: LDN is typically taken at bedtime (9 to 11 PM) for two reasons. First, opioid receptor density and endorphin production undergo circadian variation, with nocturnal peaks. Taking LDN at night to trigger the compensatory rebound during the period of highest natural opioid activity may optimize the endorphin upregulation effect. Second, vivid dreams (the most common side effect) are less disruptive when the drug is clearing in the morning hours rather than during daytime. R
Some clinicians prefer morning dosing to avoid any sleep disruption; patient response guides the choice.
ULDN dosing range: 1 nanogram to 1 microgram per day (approximately 0.001 to 1 microg per day)
ULDN requires specialized compounding and is currently used primarily as a co-administered adjunct with opioid analgesics in pain management settings. It is not typically self-administered.
Formulation: LDN requires a compounding pharmacy because no 1.5 to 4.5 mg oral formulation is commercially manufactured. Capsules are the most common form. Liquid formulations allow more precise dose titration and are available from some compounders. Topical cream formulations are used by some practitioners, particularly for localized inflammatory conditions.
Sourcing: A prescription from a licensed physician is required. Most compounding pharmacies charge approximately $30 to $60 per month for LDN. The online LDN Research Trust maintains a list of LDN-prescribing clinicians and compounding pharmacies.
Caveats And Contraindications
Cannot be used with opioid analgesics (at LDN doses): LDN's opioid receptor blockade, even partial, will precipitate withdrawal in opioid-dependent patients and reduce the efficacy of opioid pain medications. Patients on chronic opioid therapy cannot use LDN. (ULDN can be co-administered with opioids for the opposite purpose: potentiating analgesia.)
Autoimmune conditions on immunosuppressants: Because LDN modulates rather than suppresses the immune system, it may theoretically interact with immunosuppressant medications used for organ transplantation or severe autoimmune disease. This requires careful clinical assessment.
Thyroid disease: The OGF axis has been studied in the cornea, and naltrexone has been used in diabetic keratopathy. There are some reports of LDN affecting thyroid antibody levels. Thyroid medication requirements may change with LDN use; monitoring is appropriate.
Vivid dreams: The most common side effect, occurring in approximately 20 to 30% of users, particularly in the first few weeks. Changing the timing of the dose usually resolves this.
The opioid-free period required before starting: To avoid precipitating withdrawal, patients must be opioid-free for 7 to 10 days before beginning LDN. This includes tramadol, codeine, buprenorphine, and all other opioid-containing medications.
Evidence gaps: LDN has not been studied adequately in pregnancy, pediatric populations outside of Crohn's disease research, or patients with severe renal or hepatic impairment. The absence of evidence is not the same as evidence of absence, but clinical caution is warranted in these groups.
Synergies: Sauna And Cannabinoids
The mechanisms discussed throughout this post do not exist in isolation. Several widely available interventions engage the same pathways through different upstream routes, producing additive or complementary effects when combined with LDN.
Sauna And Hyperthermia
Sauna produces two effects directly relevant to LDN's mechanisms: a strong beta-endorphin surge and heat shock protein (HSP) induction that intersects with TLR4 signaling.
Beta-endorphin and the opioid rebound mechanism:
Sauna heat stress is one of the most reliably documented non-exercise triggers of beta-endorphin release. R Plasma ACTH (adrenocorticotropic hormone) and beta-endorphin both rise significantly during and after sauna exposure, driven by hypothalamic pro-opiomelanocortin (POMC) cleavage in response to thermal stress. R This is the same endogenous opioid system that LDN's rebound mechanism seeks to upregulate.
The synergy here is additive, not redundant. LDN temporarily blocks opioid receptors, triggering compensatory upregulation of receptor density and endorphin production during the 18 to 20 hours of no blockade. Sauna, used during those clear hours, delivers an additional beta-endorphin bolus to a system whose receptors have been upregulated by LDN. The subjective and analgesic consequences of this endorphin surge are likely amplified by the enhanced receptor sensitivity LDN produces. R
A compelling observation in the hyperthermia literature: heroin, cocaine, and alcohol addicts show absent or blunted beta-endorphin responses to sauna heat stress. The chronic overstimulation of opioid neurotransmission from these substances suppresses the endogenous opioid response to thermal stress. LDN works in the opposite direction, restoring and enhancing endogenous opioid tone. R For patients recovering from opioid dependence, this creates a plausible case for sauna as a lifestyle intervention that exercises and strengthens the same opioid signaling pathway that LDN is rebuilding.
Heat shock proteins and TLR4:
Sauna and other heat exposures induce the intracellular heat shock protein 70 (HSP70) family, the most studied stress-induced molecular chaperone. R Passive heating (40 degrees C water immersion) produces extracellular HSP70 increases comparable to moderate exercise in both lean and overweight men, confirming that the sauna-level heat stress is a sufficient trigger. R
The HSP70-TLR4 relationship is not simple and must be understood with nuance:
Intracellular HSP70, induced by heat stress and acting within the cell, is anti-inflammatory. It inhibits NF-kB activation, reduces iNOS (inducible nitric oxide synthase) expression and NO production in microglia, and can suppress LPS-induced microglial activation. R When HSP70 is knocked down by siRNA in microglial cell lines, the anti-inflammatory effect of HSP90 inhibitors (which upregulate HSP70) is lost, confirming that intracellular HSP70 mediates the anti-inflammatory protection. R
HSP70 also induces IL-10 expression through TLR2 and TLR4-dependent mechanisms, promoting an anti-inflammatory cytokine environment. R Blocking HSP90 (which is constitutively active and promotes inflammation) activates heat shock factor 1 (HSF1), upregulates HSP70, and downregulates NF-kB activation, producing a net anti-inflammatory state. R
The complication:
Extracellular HSP70, released from necrotic cells or actively secreted, can act as a damage-associated molecular pattern (DAMP) via TLR2 and TLR4 on immune cells, potentially triggering pro-inflammatory signaling. This dual nature means the localization and form of HSP70 matters: intracellular HSP70 protects and inhibits neuroinflammation; extracellular HSP70 from damaged tissue can amplify it. R
In the context of sauna use in healthy individuals, the dominant effect appears to be beneficial intracellular HSP70 induction rather than pathological extracellular release from cell death. Regular sauna sessions increase intracellular HSP70 progressively with repeated exposure, producing the metabolic and anti-inflammatory adaptations seen in regular sauna users. R
Practical synergy:
LDN dampens TLR4 signaling on microglia via direct TRIF-IRF3 antagonism. Regular sauna supports this via intracellular HSP70 induction that independently reduces NF-kB-driven microglial activation through a distinct molecular pathway. The two interventions act on the same inflammatory output (microglial over-activation and TLR4-driven cytokine release) through complementary upstream mechanisms. They are not redundant; they are mechanistically distinct routes to the same goal.
Timing note: Sauna sessions in the evening, after LDN has been taken and is beginning to clear, may optimize the beta-endorphin interaction. LDN is often taken at bedtime; an early evening sauna (8 to 9 PM) falls during the peak hours of LDN activity and the early clearing phase, potentially combining the direct endorphin release from heat stress with the beginning of the receptor rebound window.
Cannabinoids: THC, CBD, And The CB2-TLR4-Opioid Triangle
The endocannabinoid system and the opioid system are deeply intertwined, both anatomically and pharmacologically. Cannabinoids engage all three of LDN's relevant pathways through distinct receptor mechanisms.
CB2 receptors, microglia, and TLR4:
Microglial cells express functional cannabinoid receptor type 2 (CB2R), and CB2R activation drives a switch from M1 (pro-inflammatory) to M2 (anti-inflammatory) microglial phenotype. R
Activation of microglial CB2R inhibits ERK1/2 phosphorylation, reducing iNOS production and pro-inflammatory cytokine release in multiple neuroinflammatory conditions. R
Beta-caryophyllene, a dietary terpene found in cannabis (and black pepper, cloves, and hops) that selectively activates CB2R, independently inhibits the CD14/TLR4/MD-2 signaling complex, reducing IL-1beta, IL-6, IL-8, and TNF-alpha production. R This makes beta-caryophyllene a non-psychoactive CB2 agonist that directly overlaps with LDN's TLR4 antagonism.
CBD and TLR4-NF-kB signaling:
Cannabidiol (CBD) inhibits LPS-induced TLR4 activation in microglia through multiple mechanisms: It partially reverses IRAK-1 degradation and IkB degradation, reducing NF-kB subunit phosphorylation and nuclear translocation. R It reduces proinflammatory STAT1 activation while strengthening anti-inflammatory STAT3 signaling, shifting the cytokine profile toward IL-10 dominance. R CBD also activates CB2R and increases endogenous anandamide (AEA) and 2-arachidonoylglycerol (2-AG) levels by inhibiting their enzymatic degradation, prolonging endocannabinoid tone. R
In a rodent neuropathic pain model, CBD prevented paclitaxel-induced neuropathic pain, inhibited spinal TLR4 and Iba1 (microglial marker) expression, increased spinal endocannabinoid levels, and reduced pro-inflammatory cytokines, all via a CB2-dependent mechanism. R
The mechanistic overlap with LDN is direct: both LDN and CBD target TLR4 signaling on microglia, but through distinct molecular mechanisms (TRIF-IRF3 antagonism for LDN; NF-kB and STAT pathway modulation for CBD), making combination use mechanistically complementary rather than redundant.
THC, CB1R, and endogenous opioid release:
THC (delta-9-tetrahydrocannabinol) produces analgesia through both CB1R-mediated mechanisms and, notably, through the release of endogenous opioid peptides. Several studies show that the antihyperalgesic effects of FAAH (fatty acid amide hydrolase) inhibitors (which raise anandamide) are blocked by naloxone and kappa opioid receptor (KOR) antagonists, confirming that cannabinoid-mediated analgesia is partly mediated through endogenous opioid release. R
CB2R activation also releases beta-endorphin as a mechanism of peripheral antinociception. R Delta-9-THC releases dynorphin (an endogenous kappa opioid peptide), and this release is involved in both its analgesic effects and its contribution to tolerance resistance in combination dosing. R
This means THC, working through the endocannabinoid system, releases endogenous opioids that then act on the same opioid receptors that LDN's rebound mechanism has upregulated. If LDN has increased mu-opioid receptor density and endorphin sensitivity, THC-triggered endorphin release would produce a larger signal on those upregulated receptors.
THC-opioid synergy and the opioid-sparing effect:
The most clinically relevant cannabinoid-opioid synergy is the opioid-sparing effect. A systematic review and meta-analysis of preclinical studies found that the median effective dose (ED50) of morphine co-administered with THC was 3.5 to 3.6 times lower than the ED50 of morphine alone, a greater-than-additive (synergistic) effect. R
In a human clinical study of chronic pain patients maintained on sustained-release morphine or oxycodone, inhaled cannabis reduced pain by an average of 27% without changing opioid plasma concentrations, suggesting a pharmacodynamic rather than pharmacokinetic interaction. R
For patients using LDN specifically for its endorphin-rebound mechanism (rather than for TLR4 blockade), this raises an important consideration: LDN works by briefly blocking opioid receptors to trigger upregulation. Using THC (which releases endogenous opioids) during the 4 to 6 hour period when LDN is actively blocking receptors would compete with the blockade mechanism. Using THC during the 18 to 20 hour clear period allows the upregulated opioid system to receive the additional endogenous opioid signal from cannabinoid-driven release.
CB2R agonists and morphine tolerance:
The TLR4-NLRP3 inflammasome pathway is a central driver of morphine tolerance. Repeated morphine administration increases spinal TLR4 expression, activates NLRP3 (NOD-like receptor family pyrin domain-containing protein 3), and drives neuroinflammatory tolerance. R CB2R activation reduces microglial activation and suppresses this TLR4-NLRP3 tolerance cascade, delaying or attenuating morphine tolerance development. R LDN and CB2R agonists (or full-spectrum cannabis containing beta-caryophyllene and CBD) target morphine tolerance through converging anti-TLR4 mechanisms, both acting on the neuroinflammatory component of tolerance.
The important caveat on THC and LDN together:
LDN cannot be co-administered with exogenous opioids because it will precipitate withdrawal and block opioid analgesic effects. Endogenous opioids released by THC and other cannabinoids are not the same as exogenous opioid medications and are not present at high enough concentrations to precipitate opioid withdrawal in the LDN context. However, timing still matters. Using THC during the 4 to 6 hour window of active LDN opioid receptor blockade will blunt the opioid effects of the endogenous peptides THC releases. This is not dangerous, but it is pharmacologically wasteful. Using THC (or any cannabis product expected to act partly through endogenous opioid release) during the clear hours maximizes the synergistic potential. R
Summary table: synergy mechanism map
| Intervention | TLR4 / Microglial | Endorphin Rebound | OGF Axis | Notes |
|---|---|---|---|---|
| LDN (1 to 5 mg) | Direct TRIF-IRF3 antagonism | Indirect: brief receptor blockade triggers upregulation | Intermittent OGFr blockade drives upregulation | Primary mechanisms |
| Sauna / hyperthermia | Intracellular HSP70 reduces NF-kB and iNOS | Direct beta-endorphin surge via POMC/hypothalamus | Unknown | Use sauna during LDN clear hours for additive endorphin effects |
| CBD | TLR4-NF-kB and STAT1 downregulation via CB2R | Raises endocannabinoid tone (indirect) | Unknown | Mechanistically complementary to LDN on TLR4 |
| Beta-caryophyllene | Direct CD14/TLR4/MD-2 complex inhibition via CB2R | Peripheral beta-endorphin release via CB2R | Unknown | Non-psychoactive; terpene found in cannabis, black pepper |
| THC | CB2R shifts microglia M1 to M2 | Releases dynorphin and endorphins (CB1R-dependent) | Unknown | Use during LDN clear hours; do not use during 4 to 6 hour blockade window |
| CB2-selective agonists | Suppresses TLR4-NLRP3 tolerance cascade | Beta-endorphin release | Unknown | Most directly synergistic with LDN mechanisms; no psychoactivity |
Mechanisms Of Action
Simple:
- At 1 to 5 mg per day (LDN), naltrexone acts primarily as a TLR4 antagonist on microglial cells, blocking the TRIF-IRF3 inflammatory signaling pathway that produces nitric oxide, TNF-alpha, and reactive oxygen species.
- This shifts activated microglia from an M1 (pro-inflammatory) to an M2 (anti-inflammatory) phenotype, reducing the neuroinflammation driving chronic pain, fatigue, and immune dysregulation.
- Simultaneously, the brief 4 to 6 hour opioid receptor blockade from LDN triggers a homeostatic upregulation of endogenous opioids (beta-endorphin, met-enkephalin) and opioid receptors during the 18 to 20 hours after the drug clears.
- Enhanced endorphin tone improves analgesia, mood, NK cell activity, and immune regulation throughout most of the day.
- LDN also upregulates the OGF-OGFr axis (met-enkephalin / zeta receptor), which slows pathological cell proliferation in cancer, reduces autoreactive T and B lymphocyte proliferation, and may reduce the disease activity of autoimmune conditions.
- At below 1 microgram per day (ULDN), the operative mechanism is filamin A binding, which prevents the Gi/o to Gs G protein coupling switch in the mu-opioid receptor that underlies opioid tolerance, hyperalgesia, and dependence.
- The TLR4 and opioid receptor mechanisms are dose-independent: TLR4 binding dominates at 1 to 5 mg, while opioid receptor rebound requires brief blockade that clears; ULDN operates in a dose window so narrow (picomolar) that slightly higher doses abolish the effect.
Advanced:
- TLR4 TRIF-IRF3 pathway biology: TLR4 signal transduction occurs through two intracellular adapters. MyD88 recruits IRAK1/4 to activate the NF-kB and MAPK cascades producing early cytokines (IL-1beta, IL-6, TNF-alpha). TRIF is recruited independently, activating TBK1 and IKKepsilon which phosphorylate IRF3, driving IRF3 homodimerization, nuclear translocation, and transcription of IFN-beta and iNOS. Both (+)- and (-)-naltrexone specifically block the TRIF-IRF3 arm of TLR4 signaling, inhibiting IRF3 phosphorylation and nuclear translocation without significantly disrupting MyD88-NF-kB signaling. This biased antagonism reduces the delayed and sustained inflammatory output (NO, IFN-beta, ROS) associated with chronic microglial activation, while preserving some acute innate immune responsiveness through the MyD88 pathway. R
- OGF-OGFr nuclear signaling: OGF ([Met5]-enkephalin) enters cells via clathrin-mediated endocytosis in a temperature-dependent manner, appearing in the cytoplasm within 15 minutes and reaching both cytoplasm and nucleus within 30 minutes of exposure. OGFr, located on the outer nuclear envelope, contains nuclear localization signals that interact with karyopherin-beta and Ran GTPase for nuclear import. The OGF-OGFr complex accumulates in the paranuclear cytoplasm and translocates into the nucleus where it upregulates p16 (CDKN2A) and p21 (CDKN1A) cyclin-dependent kinase inhibitors. These CDKIs inhibit cyclin D/CDK4 and cyclin E/CDK2 complexes, arresting cells at the G1/S checkpoint. Because OGFr is pharmacologically distinct from mu, delta, and kappa receptors in molecular structure and receptor pharmacology (sharing only receptor blocker sensitivity), OGF's growth regulatory action is separable from classical opioid analgesia. R
- Filamin A picomolar binding site: FLNA2561-2565 (the pentapeptide KWLAM within the C-terminal domain of FLNA) is the high-affinity binding site for naloxone and naltrexone at approximately 4 pM affinity. This site is structurally distinct from opioid receptor binding sites and is intracellular, explaining why ultra-low concentrations that cannot occupy the far more numerous opioid receptors can still produce biological effects. When naltrexone binds FLNA at this site, it prevents the conformational change in the MOR-FLNA complex that permits Gs coupling. The MOR-Gs interaction is sterically blocked; adenylyl cyclase activation is prevented; cAMP upregulation and the downstream CREB (cAMP response element-binding protein) excitatory signaling cascade are attenuated. The second, lower-affinity FLNA binding site at approximately 834 pM appears to disrupt rather than reinforce the effect of the high-affinity site, producing the paradoxical loss of effect at slightly higher concentrations. R
Genetics
OPRM1 (mu-opioid receptor gene) A118G (rs1799971): The most studied variant in opioid pharmacogenomics. The G allele (Asp40 or N40D) reduces opioid receptor expression and alters receptor signaling. G allele carriers have reduced endogenous opioid tone at baseline, reduced response to opioid analgesics, and altered alcohol reward signaling. For LDN specifically, the opioid rebound mechanism depends on a functional MOR system to upregulate; A118G status may predict the magnitude of the endorphin rebound response. G allele carriers may require different LDN doses or may respond preferentially to LDN's TLR4 mechanism rather than the opioid rebound mechanism. R
TLR4 variants: TLR4 Asp299Gly (rs4986790) and Thr399Ile (rs4986791) are common variants that reduce TLR4 signaling responsiveness by impairing TLR4 dimerization and ligand recognition. Carriers of these variants have blunted TLR4-mediated innate immune responses and may also have reduced responsiveness to naltrexone's TLR4 antagonism if the receptor itself is already hypo-functional. On the other hand, in conditions driven by TLR4 over-activation, hyperactive TLR4 variants may predict greater LDN benefit. This pharmacogenomic relationship has not been directly studied in LDN trials. R
COMT Val158Met (rs4680): Catechol-O-methyltransferase (COMT) metabolizes catecholamines and affects pain sensitivity, stress response, and opioid receptor availability. Met/Met homozygotes have lower COMT activity, resulting in higher dopamine and norepinephrine levels, reduced mu-opioid receptor binding potential in the brain, and generally higher pain sensitivity. Met/Met individuals may have a different baseline opioid tone and a different response to the opioid rebound mechanism of LDN. COMT genotype is one of the most studied predictors of fibromyalgia susceptibility and pain sensitivity, making it a plausible predictor of LDN response in fibromyalgia specifically.
More Research
- TLR4 and opioid tolerance: The same TLR4 on microglia that LDN blocks is also activated by opioids themselves. This is not a well-known fact but has significant implications. Opioids activate TLR4 via a non-stereoselective interaction (similar to naltrexone's TLR4 binding), triggering microglial inflammation that contributes to opioid-induced hyperalgesia (OIH) and tolerance. LDN's TLR4 blockade during the 4 to 6 hours of activity may therefore partially suppress the microglial activation that opioids themselves are driving, which would explain why ULDN co-administration reduces tolerance even beyond the FLNA mechanism. R
- Mast cells and TLR4: TLR4 receptors are expressed on mast cells as well as microglia. Mast cell activation via TLR4 contributes to several conditions with possible LDN benefit, including mast cell activation syndrome (MCAS), migraine, and dysautonomia. LDN's TLR4 antagonism on mast cells represents a peripheral immune mechanism separate from the CNS microglial mechanism and is particularly relevant for patients with MCAS-driven symptom patterns. R
- The very low dose range (VLDN, 1 microg to 1 mg): A dose range between ULDN and LDN has been used primarily in opioid-weaning protocols as a methadone taper adjunct. This range reduces opioid withdrawal severity and improves the tolerability of dose reduction. The mechanism is likely a blend of partial FLNA engagement at the lower end and early TLR4 engagement at the upper end, though this has not been formally characterized. R
- LDN and the gut microbiome: TLR4 activation in the gut wall is driven substantially by dysbiosis-derived LPS (endotoxin) from leaky gram-negative bacteria. If LDN is partly reducing TLR4 signaling triggered by gut LPS, then optimizing gut barrier integrity through resistant starch, butyrate-raising strategies, and microbiome restoration would be synergistic with LDN. Reducing the LPS load in circulation would reduce TLR4 activation systemically, and LDN would dampen residual TLR4 signaling from whatever LPS still crosses the gut wall. This is a clinically important combination strategy for patients whose neuroinflammation or chronic pain has a gut dysbiosis component.
- The opioid-independent effects and what dextro-naltrexone confirmed: The clearest evidence that LDN's pain and anti-inflammatory effects are not primarily opioid-mediated comes from the fact that dextrorotatory (optically right-handed) naltrexone, which cannot engage opioid receptors at all, still produces neuroprotective and anti-inflammatory effects. This finding, combined with the TRIF-IRF3 selectivity demonstrated in microglial cell lines devoid of opioid receptors, makes TLR4 the most mechanistically sound explanation for LDN's efficacy in conditions defined by neuroinflammation. R
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Quercetin
500mg 2x/day
Vitamin D3 + K2
5000 IU + 200mcg/day
DAO Enzyme
1 cap before meals