All Ten Human Toll-Like Receptors: What Each One Senses, How It Signals, And Where It Goes Wrong
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.

The innate immune system does not respond to danger randomly.
It operates through a precisely organized family of pattern recognition receptors that each detect a specific class of molecular signature associated with infection or tissue damage.
Toll-like receptors (TLRs) are the most studied of these sensors.
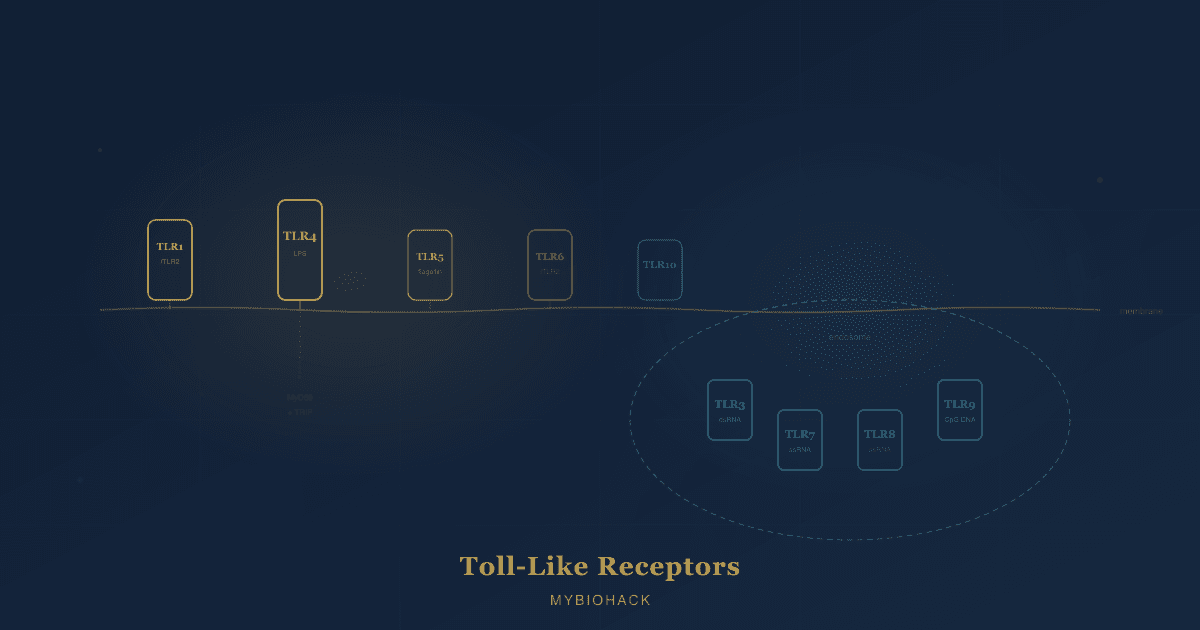
Humans express ten of them (TLR1 through TLR10), each positioned either on the cell surface or inside endosomal compartments, each recognizing a distinct set of ligands, and each coupling to partially overlapping but distinguishable downstream signaling cascades. R
This post covers all ten human TLRs: their structure, the ligands they detect, the cells that express them, the signaling pathways they activate, and the diseases that emerge when this system malfunctions.
Shared Architecture: How TLRs Are Built
All ten human TLRs share a common three-domain architecture. R
The extracellular domain is composed of leucine-rich repeat (LRR) motifs arranged in a characteristic horseshoe shape.
LRRs form the ligand-binding surface and determine the specificity of each TLR for its particular pathogen-associated molecular pattern (PAMP) or damage-associated molecular pattern (DAMP).
The transmembrane domain anchors the receptor in either the plasma membrane (for cell surface TLRs) or the endosomal membrane (for intracellular TLRs).
The cytoplasmic Toll/IL-1 receptor (TIR) domain is the signaling interface.
After ligand-induced dimerization of the extracellular domain, the TIR domains of the two receptor subunits come together and recruit intracellular adapter proteins that initiate downstream signaling cascades. R
Most TLRs function as homodimers (one TLR pairing with an identical partner).
TLR2 is the major exception: it obligately functions as a heterodimer, pairing with TLR1, TLR6, or TLR10 to expand its ligand recognition range. R
Chaperone system for TLR trafficking:
TLRs are synthesized in the endoplasmic reticulum (ER) and require dedicated chaperone proteins to fold correctly and traffic to the correct cellular compartment.
gp96 (an ER-resident member of the HSP90 family) is the universal chaperone for most TLRs.
PRAT4A (protein associated with TLR4) regulates ER exit and trafficking for TLR1, TLR2, TLR4, TLR7, and TLR9.
UNC93B1 is a 12-pass transmembrane ER protein specifically required for all nucleic acid-sensing TLRs (TLR3, TLR7, TLR8, TLR9) to exit the ER and traffic to endosomes. R
Loss of UNC93B1 function causes susceptibility to herpes simplex encephalitis in humans.
Two Signaling Pathways: MyD88 And TRIF
The downstream response of virtually every human TLR flows through one or both of two intracellular adapter proteins.
MyD88 (myeloid differentiation primary response 88) is used by all human TLRs except TLR3.
After receptor activation, the TIR domain recruits either TIRAP (also called MAL, a bridging adapter) or directly engages MyD88.
MyD88 then recruits IRAK4 (IL-1 receptor-associated kinase 4), which activates IRAK1 and IRAK2, which in turn activate TRAF6 (TNF receptor-associated factor 6).
TRAF6 activates the TAK1 kinase complex, which phosphorylates IκB, releasing NF-κB to translocate to the nucleus and drive transcription of pro-inflammatory cytokines (IL-1β, IL-6, IL-8, TNF-α) and chemokines. R
In plasmacytoid dendritic cells (pDCs), the MyD88 pathway additionally recruits TRAF3 and IRAK1, leading to direct phosphorylation of IRF7 and the production of massive amounts of type I interferons (IFN-α/β). R
TRIF (TIR-domain-containing adapter-inducing interferon-β) is the second adapter, used exclusively by TLR3 (alone) and TLR4 (in its endosomal phase, in concert with the bridging adapter TRAM).
TRIF activates TRAF3, which recruits TBK1 (TANK-binding kinase 1) and IKKε.
TBK1 and IKKε phosphorylate IRF3 (and to a lesser extent IRF7), driving IRF3 dimerization and nuclear translocation to induce type I interferons (primarily IFN-β) and interferon-stimulated genes (ISGs). R
TLR4 is unique in being the only human TLR that uses all four adapters (TIRAP, MyD88, TRAM, TRIF), activating both pathways. R
Outputs by pathway:
- MyD88: pro-inflammatory cytokines (NF-κB driven), IFN-α (pDC-specific, via IRF7)
- TRIF: type I interferons primarily (IFN-β via IRF3), delayed NF-κB activation via RIP1 kinase
The Two Groups: Cell Surface vs Endosomal
Human TLRs partition into two distinct subcellular locations based on the class of ligands they detect.
Cell surface TLRs (TLR1, TLR2, TLR4, TLR5, TLR6, TLR10) are positioned on the plasma membrane where they survey the extracellular environment for bacterial membrane components: lipids, lipoproteins, and proteins. R
Endosomal TLRs (TLR3, TLR7, TLR8, TLR9) reside within intracellular endosomal and lysosomal compartments where they detect microbial nucleic acids (double-stranded RNA, single-stranded RNA, and unmethylated CpG DNA) after the pathogen has been endocytosed and its nucleic acids are released into the acidic endosomal lumen. R
The endosomal localization of nucleic acid-sensing TLRs is a critical self-tolerance mechanism.
By restricting access to acidic endosomal compartments, the immune system ensures that extracellular self-DNA and RNA in the circulation do not normally access TLR7 or TLR9, preventing inadvertent activation by host nucleic acids.
When this compartmentalization is disrupted (as in autoimmune disease), self-nucleic acids can gain access to these intracellular sensors and trigger autoimmune responses. R
Additionally, all endosomal nucleic acid-sensing TLRs undergo proteolytic cleavage by cathepsins (B, S, L, H, K) within the acidic endosomal lumen to achieve their active functional form.
This proteolytic processing provides an additional layer of compartment-specific activation control. R
TLR1: The Triacyl Lipopeptide Sensor
Location: Cell surface
Dimerization: Obligate heterodimer with TLR2
Signaling: MyD88 only
What TLR1 recognizes:
TLR1 does not function alone.
It serves as the co-receptor that confers triacyl lipopeptide specificity to the TLR2/TLR1 heterodimer. R
Triacylated lipopeptides are the dominant lipid-modified proteins on gram-positive bacteria (most commonly) and mycobacteria.
They possess a unique N-terminal acyl modification with three fatty acid chains.
Structural crystallography explains the ligand discrimination mechanism precisely: the TLR1 ectodomain contains a hydrophobic channel that accommodates the amide-linked third acyl chain of triacylated lipopeptides.
TLR6 (the competing TLR2 co-receptor) has the same channel blocked by bulky amino acid residues, making it structurally incapable of binding the third acyl chain and therefore selective for diacylated lipopeptides instead. R
Primary cells and tissues:
TLR1 is broadly expressed on monocytes, macrophages, dendritic cells, neutrophils, and T cells.
TLR1 expression on CD4+ T cells is upregulated upon T cell activation, allowing lipopeptide-induced direct T cell costimulation independent of antigen-presenting cells (APCs). R
Key ligands:
- Pam3CSK4 (synthetic triacylated lipopeptide; standard TLR2/1 research agonist)
- Mycobacterium tuberculosis lipoproteins LprG and LpqH
- Borrelia burgdorferi outer surface proteins (OspA, OspB)
- Staphylococcal lipoproteins
Disease relevance:
TLR1 polymorphisms are among the best-characterized contributors to tuberculosis susceptibility in humans.
The T1805G (I602S) polymorphism reduces TLR1 surface expression and lipopeptide-induced cytokine secretion, and is associated with increased susceptibility to tuberculosis and leprosy in multiple human populations. R
Decreased TLR1/TLR6 gene expression is also associated with reduced BCG-induced cytokine production in South African infants, connecting TLR1 to vaccine efficacy. R
In plasmacytoid dendritic cells, TLR1 (but not TLR2) blockade abolishes IL-6 and TNF-α production while TLR2 (but not TLR1) blockade suppresses type I IFN secretion, demonstrating that the two subunits of the TLR1/2 heterodimer differentially control distinct inflammatory outputs. R
TLR2: The Bacterial Lipid Hub
Location: Cell surface (also traffics to endosomes in some inflammatory monocytes)
Dimerization: Heterodimer with TLR1 (triacyl), TLR6 (diacyl), or TLR10
Signaling: MyD88 only (plus limited type I IFN in specialized contexts via endosomal trafficking)
What TLR2 recognizes:
TLR2 is the most ligand-promiscuous TLR.
It recognizes an exceptionally broad range of bacterial, fungal, viral, and parasitic components, with its specificity determined by its dimerization partner.
The major TLR2 ligand class is bacterial lipoproteins, which are ubiquitous across virtually all bacteria.
Lipoproteins present a unique N-terminal lipo-amino acid (N-acyl-S-diacylglycerol cysteine) and are the molecular signature of bacterial membranes that TLR2 is specifically designed to detect. R
TLR2/TLR1 heterodimer: triacylated lipoproteins (gram-positive bacteria, mycobacteria)
TLR2/TLR6 heterodimer: diacylated lipoproteins (mycoplasma, gram-positive), lipoteichoic acid (LTA), MALP-2 (mycoplasmal macrophage-activating lipopeptide-2), zymosan (fungi), phospholipomannan (Candida albicans)
TLR2 homodimer or TLR2 with other partners: peptidoglycans, lipoarabinomannan (mycobacteria), GPI-mucin from Trypanosoma cruzi
Additional TLR2 ligands include:
- Hepatitis C virus core protein
- Measles virus hemagglutinin
- Herpes simplex virus glycoprotein B
- HMGB1 (endogenous alarmin released from necrotic cells)
- Heat shock proteins (HSP60, HSP70) from damaged tissue R
Primary cells and tissues:
TLR2 is expressed on monocytes, macrophages, dendritic cells, neutrophils, mast cells, B cells (low), and some T cell subsets.
Expression is upregulated during inflammation and infection.
Disease relevance:
TLR2 is the primary innate immune sensor for Mycobacterium tuberculosis lipoproteins.
Over 48 to 100 lipoproteins encoded in the Mtb genome are TLR2 ligands.
The 19-kDa lipoprotein (LpqH) was the first Mtb ligand shown to signal through TLR2.
TLR2 activation by Mtb lipoproteins both drives antimicrobial responses (early infection) and impairs IFN-γ responsiveness and MHC class II antigen presentation (chronic infection), suggesting that Mtb exploits sustained TLR2 signaling to persist in macrophages. R
TLR2 is the dominant receptor for Borrelia burgdorferi (Lyme disease) outer surface lipoproteins, with TLR2/TLR6 cooperating to recognize OspA-L. R
Elevated TLR2 expression on neutrophils from Lyme disease patients correlates with elevated IL-6 production, consistent with TLR2-driven inflammatory disease. R
TLR2 polymorphisms (including the 597C variant in Vietnamese populations) are associated with tuberculosis susceptibility.
TLR2 also recognizes endogenous DAMPs including HMGB1 and heat shock proteins, connecting it to sterile inflammatory conditions including atherosclerosis and ischemia-reperfusion injury.
TLR3: The Double-Stranded RNA Detector
Location: Endosomal
Dimerization: Homodimer
Signaling: TRIF only (the only human TLR that is entirely MyD88-independent)
What TLR3 recognizes:
TLR3 detects double-stranded RNA (dsRNA), which is produced as an intermediate during viral replication by virtually all RNA viruses (dsRNA viruses, and positive and negative-sense ssRNA viruses produce dsRNA replication intermediates). R
TLR3 also recognizes:
- Poly(I:C) (polyinosinic-polycytidylic acid), the synthetic dsRNA analog used in research and as a vaccine adjuvant
- Small interfering RNAs (siRNAs) and other endogenous RNAs released from damaged cells
- mRNA from necrotic cells
The TLR3 activation mechanism is structurally elegant: dsRNA binds along the concave dimerization interface of the TLR3 homodimer, contacting two distinct sites on each protomer (one near the N-terminus and one near the C-terminus of the solenoid), creating a 2:2 receptor-RNA complex that brings the TIR domains together for signaling. R
Signaling output:
TLR3 exclusively uses TRIF, making it the only TLR that does not activate the MyD88-NF-κB pathway.
TRIF activation drives TBK1 and IKKε-mediated phosphorylation of IRF3, producing IFN-β and ISGs (interferon-stimulated genes) as the primary output.
TRIF also activates NF-κB via TRAF6 and RIP1, producing delayed inflammatory cytokines. R
Primary cells and tissues:
TLR3 is predominantly expressed in myeloid dendritic cells (mDCs), macrophages, fibroblasts, and epithelial cells.
It is notably absent from B cells and has restricted expression in plasmacytoid dendritic cells (unlike TLR7 and TLR9). R
Disease relevance:
Herpes simplex encephalitis (HSE): TLR3 loss-of-function mutations are one of the few clearly established Mendelian genetic causes of susceptibility to a specific viral infection in humans.
Children with TLR3 pathway defects (including mutations in TLR3, UNC93B1, TRIF, TRAF3, TBK1, and IRF3) are selectively vulnerable to HSV-1 encephalitis, while remaining resistant to other herpesviruses and common infections.
This demonstrates that the TLR3-TRIF-IRF3 pathway is non-redundantly required for antiviral IFN production in the central nervous system against HSV-1. R
SLE (systemic lupus erythematosus): TLR3 contributes to lupus nephritis progression via activation of glomerular mesangial cells and APCs by dsRNA-containing self-material, though it does not drive anti-dsDNA antibody production (that role belongs to TLR9). R
HCV evasion: Hepatitis C virus (HCV) NS3/4A protease cleaves TRIF, blocking TLR3-dependent IFN-β production in hepatocytes and enabling viral persistence. R
TLR4: The Gram-Negative Sentinel
Location: Cell surface (then endocytosed to endosome upon activation)
Dimerization: Homodimer, with essential co-receptors MD-2 and CD14
Signaling: Both MyD88 (cell surface phase) and TRIF (endosomal phase); the only TLR using all four adapters
What TLR4 recognizes:
The canonical TLR4 ligand is lipopolysaccharide (LPS), the major outer membrane component of gram-negative bacteria.
LPS recognition requires a multi-component co-receptor complex: LPS-binding protein (LBP) extracts LPS monomers from bacterial outer membranes, CD14 delivers LPS to MD-2, and MD-2 presents the lipid A moiety of LPS to the TLR4 ectodomain, allowing dimerization and signaling. R
TLR4 is the most important receptor for sensing gram-negative sepsis.
Additional TLR4 ligands include:
- Lipid A (the bioactive component of LPS)
- Taxol (plant-derived, activates mouse but not human TLR4)
- Respiratory syncytial virus (RSV) fusion protein
- HMGB1 (high mobility group box 1, an endogenous DAMP released during cell death)
- Fibronectin, fibrinogen (endogenous matrix proteins upregulated during injury)
- Heat shock proteins (HSP60, HSP70) from stressed cells
- Saturated fatty acids (palmitate, stearate)
- Oxidized LDL (oxLDL)
- Amyloid beta (linking TLR4 to Alzheimer's disease pathology)
- Opioids including morphine (at the MD-2 site, non-stereoselectively, contributing to opioid tolerance) R
Dual-phase signaling:
At the cell surface, TLR4-MD-2-LPS complex recruits TIRAP and MyD88, activating NF-κB and MAPK (early phase), producing pro-inflammatory cytokines.
After signaling, the complex is endocytosed and in the endosomal lumen recruits TRAM and TRIF, activating TBK1-IRF3 and producing IFN-β (late phase).
Both phases are required for full LPS-induced inflammatory gene expression. R
Primary cells and tissues:
TLR4 is highly expressed on monocytes, macrophages, dendritic cells, neutrophils, endothelial cells, and intestinal epithelial cells.
Notably, human B cells lack TLR4, unlike mouse B cells. R
Disease relevance:
Sepsis: Gram-negative sepsis is the prototypical TLR4 pathology.
Excessive TLR4 activation by systemic LPS drives a cytokine storm producing septic shock.
Multiple failed sepsis trials have targeted TLR4; eritoran (a TLR4 antagonist) failed in phase III clinical trials despite strong preclinical data, highlighting the complexity of innate immune targeting in critical illness.
Atherosclerosis: TLR4 activation by oxLDL and saturated fatty acids in arterial macrophages drives pro-inflammatory foam cell formation.
High-fat diets activate TLR4 by raising circulating saturated fatty acids (palmitate directly binds MD-2) and by increasing gut permeability and LPS leakage, connecting metabolic syndrome to TLR4-driven arterial inflammation. R
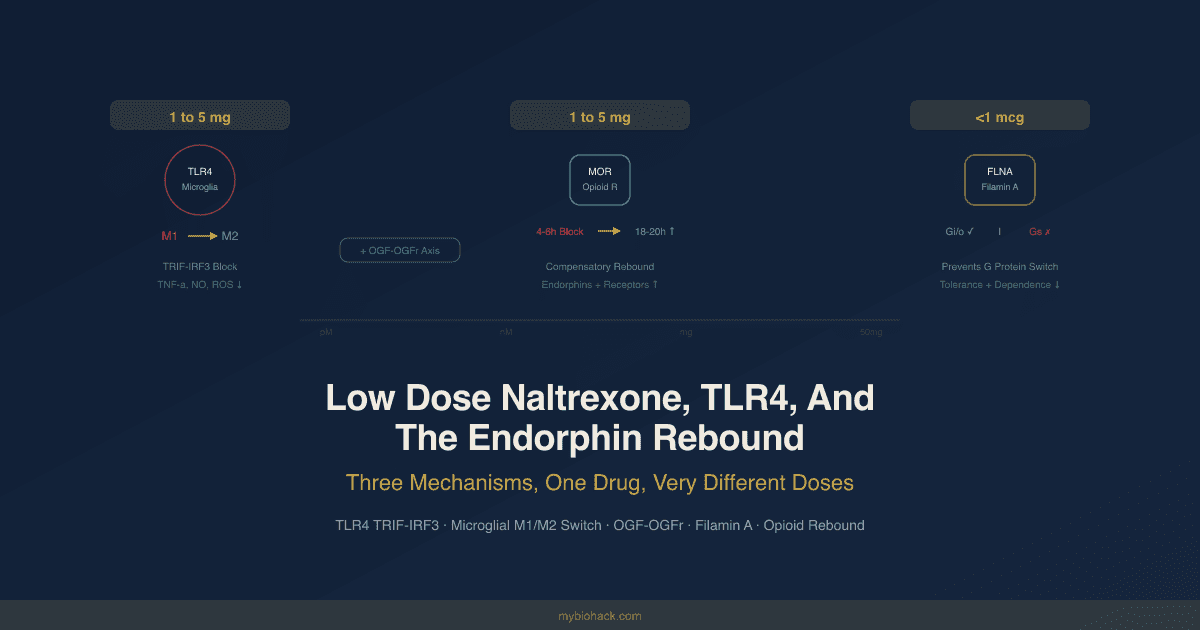
Chronic pain and opioid tolerance: Morphine, fentanyl, and other opioids activate TLR4 via the MD-2 binding site in a non-stereoselective fashion (both (+) and (-) isomers activate TLR4, unlike classical opioid receptors which only respond to (-) isomers).
Repeated morphine administration increases spinal TLR4 expression, driving microglial activation and the NLRP3 inflammasome, which are key drivers of opioid-induced hyperalgesia and tolerance. R
Low dose naltrexone (LDN) blocks this TLR4-TRIF-IRF3 axis to reduce neuroinflammation.
Neurodegeneration: Amyloid beta plaques activate TLR4 on microglia, contributing to the chronic neuroinflammatory state of Alzheimer's disease. R
TLR4 variants: Asp299Gly (rs4986790) and Thr399Ile (rs4986791) reduce LPS responsiveness by impairing TLR4-MD-2 dimerization.
These variants are associated with reduced susceptibility to gram-negative sepsis but increased susceptibility to certain intracellular infections.
TLR5: The Flagellin Receptor
Location: Cell surface
Dimerization: Homodimer
Signaling: MyD88 only
What TLR5 recognizes:
TLR5 detects flagellin, the monomeric protein subunit that polymerizes to form bacterial flagella.
Flagellin is a highly conserved structural protein across gram-negative and gram-positive motile bacteria.
TLR5 specifically recognizes a conserved domain in the D1 region of flagellin that is required for flagellar structure and is shielded within the intact flagellar filament.
This means TLR5 only detects free (monomeric) flagellin released during infection, not intact flagella on intact bacteria. R
TLR5 activation via flagellin drives robust NF-κB-dependent inflammatory cytokine production.
Primary cells and tissues:
TLR5 is expressed on intestinal epithelial cells (basolateral surface), monocytes, macrophages, dendritic cells, and NK cells.
In the gut, the basolateral positioning of TLR5 on intestinal epithelium is critically important: it ensures that flagellin from luminal bacteria accessing the basolateral compartment (which should not happen in a healthy gut) triggers an inflammatory response, while normal luminal bacterial flagellin does not constitutively activate the epithelium.
Disease relevance:
Gut homeostasis and IBD: TLR5 has a complex role in gut immunity.
In healthy individuals, TLR5 activation by invasive bacteria through the basolateral surface drives appropriate mucosal inflammation.
In inflammatory bowel disease (IBD), dysbiosis and impaired gut barrier function allow flagellin access to TLR5, contributing to chronic intestinal inflammation. R
TLR5 stop codon (rs5744168): A common loss-of-function polymorphism introducing a premature stop codon (1174C, present in roughly 7 to 10% of populations with European ancestry) creates a non-functional TLR5 protein.
Carriers produce reduced pro-inflammatory cytokines in response to flagellin.
This polymorphism is associated with protection from SLE (reduced inflammatory tone) but increased susceptibility to Legionella pneumophila infection (reduced flagellin-driven protection). R
Vaccine adjuvant: Flagellin is a highly effective TLR5 agonist vaccine adjuvant.
Flagellin-based adjuvants have demonstrated enhanced protection against Yersinia pestis, Streptococcus pneumoniae, Plasmodium falciparum, West Nile virus, influenza virus, and SARS-CoV-2 in preclinical models.
Studies also show improved vaccine responses in the elderly and enhanced longevity and healthspan in aged mice with flagellin-based immunization. R
TLR6: The Diacyl Lipopeptide Partner
Location: Cell surface
Dimerization: Obligate heterodimer with TLR2
Signaling: MyD88 only
What TLR6 recognizes:
Like TLR1, TLR6 does not function as a sensor on its own.
It is the co-receptor that confers diacyl lipopeptide specificity to the TLR2/TLR6 heterodimer. R
The structural explanation is the same as for TLR1 in reverse: TLR6's hydrophobic channel (equivalent to TLR1's triacyl chain-binding channel) is obstructed by bulky amino acid residues (phenylalanine 343 and leucine 344), making it unable to accommodate the third acyl chain of triacylated lipopeptides.
The TLR6/TLR2 heterodimer therefore recognizes only diacylated lipoproteins with two fatty acid chains.
Key ligands (recognized as TLR2/TLR6 complex):
- MALP-2 (mycoplasmal macrophage-activating lipopeptide-2): the prototypical diacylated lipopeptide
- Pam2CSK4 (synthetic diacylated lipopeptide research standard)
- Lipoteichoic acid (LTA) from gram-positive bacteria
- Borrelia burgdorferi outer surface lipoprotein A (OspA-L), requiring TLR2 and TLR6 for signaling R
- Zymosan (fungal cell wall component)
- Phospholipomannan from Candida albicans
Disease relevance:
TLR6 variants affect susceptibility to tuberculosis and the adaptive immune responses generated after BCG vaccination.
TLR1 and TLR6 deficiency together reduces BCG-induced cytokine production from T cells in South African infants, suggesting that TLR1/2 and TLR2/6 signaling shapes the vaccine-induced cellular immune response. R
TLR6 can also heterodimerize with TLR4 in response to certain endogenous ligands, contributing to sterile inflammation from tissue damage, though this is less well characterized than the TLR2/TLR6 interaction.
MALP-2 (the prototypical TLR2/TLR6 agonist) has been studied as a mucosal adjuvant for vaccines and has applications in wound healing, bone repair, and vascular regeneration through its activation of monocytes, macrophages, and endothelial cells. R
TLR7: The ssRNA Virus Alarm In pDCs
Location: Endosomal
Dimerization: Homodimer
Signaling: MyD88 (with IRF7 activation in pDCs producing massive IFN-α)
What TLR7 recognizes:
TLR7 detects single-stranded RNA (ssRNA) with a preference for guanosine- and uridine-rich sequences (GU-rich), which are common in RNA virus genomes. R
TLR7 also recognizes:
- Imidazoquinolines (imiquimod, resiquimod): small-molecule TLR7 agonists used therapeutically
- Loxoribine (guanosine analog)
- Self-RNA released from necrotic cells or in immune complexes (in autoimmune disease)
TLR7 activation requires two binding events on the homodimer: a small-molecule guanosine-like compound (or guanosine from RNA degradation products) binds one site, and a short ssRNA oligonucleotide binds a second site.
Both sites must be occupied simultaneously for receptor activation, which provides a mechanism for sensing intact RNA rather than isolated nucleosides. R
Primary cells and tissues:
TLR7 is highly expressed in plasmacytoid dendritic cells (pDCs), which are the primary producers of type I interferon in the body.
Humans also express TLR7 in B cells, where it drives autoimmune responses in SLE. R
In pDCs, TLR7 engagement signals through a MyD88 complex containing IRAK4, IRAK1, TRAF3, and TRAF6 that directly phosphorylates IRF7, producing massive IFN-α output.
Disease relevance:
Systemic lupus erythematosus: TLR7 is a central driver of SLE pathogenesis.
TLR7 overexpression or gene duplication (as seen with the Y-linked autoimmune accelerator, or Yaa, locus in mice) dramatically increases SLE severity.
In humans, gain-of-function TLR7 variants are associated with enhanced SLE susceptibility and severity, and the X-chromosome location of TLR7 (with incomplete X-inactivation in females) is a genetic basis for the strong female predominance of SLE. R
The mechanism: self-RNA from dying cells, complexed with antibodies against RNA-binding proteins (anti-Sm, anti-RNP), is internalized by autoreactive B cells via the B cell receptor (BCR) and delivered to endosomal TLR7.
TLR7 engagement provides the second signal for BCR-activated B cells, driving differentiation into autoantibody-producing plasma cells without T cell help. R
Imiquimod (therapeutic TLR7 agonist): Topical imiquimod is FDA-approved for basal cell carcinoma, genital warts (condyloma acuminata), and actinic keratosis.
It activates TLR7 (and TLR8) on local immune cells, driving IFN-α production and NK cell and CTL (cytotoxic T lymphocyte) activation to eliminate HPV-infected or malignant cells.
Paradoxically, systemic imiquimod administration in mice induces SLE-like disease, confirming that chronic systemic TLR7 activation drives autoimmunity. R
TLR8: The Myeloid ssRNA Sensor
Location: Endosomal
Dimerization: Homodimer (unique in forming a pre-formed dimer in the unliganded state)
Signaling: MyD88 only
What TLR8 recognizes:
TLR8 shares structural and functional similarity with TLR7: both detect single-stranded RNA in endosomes, require two binding events for activation (a uridine or degradation product at one site, and a ssRNA oligonucleotide at a second site), and use MyD88-NF-κB signaling. R
The key difference from TLR7 is in cellular distribution and functional output.
TLR7 is the dominant ssRNA sensor in pDCs and B cells.
TLR8 is the dominant ssRNA sensor in myeloid cells: monocytes, macrophages, myeloid dendritic cells, and neutrophils.
TLR8 also recognizes:
- Resiquimod (R848): a dual TLR7/TLR8 agonist
- GU-rich ssRNA oligonucleotides
- Degradation products of bacterial RNA
A structural distinction:
Unlike TLR7 and TLR9, which are monomeric in their unliganded state, TLR8 exists as a pre-formed dimer before ligand binding.
This pre-formed dimer undergoes a structural reorganization upon ligand binding that brings the TIR domains together for signaling. R
Disease relevance:
SLE balance with TLR7: TLR8 has an important immunoregulatory role in the context of lupus.
In mouse SLE models, absence of TLR8 (and TLR9) exacerbates TLR7-driven disease, suggesting that TLR8 and TLR9 compete with TLR7 for the endosomal trafficking chaperone UNC93B1, limiting TLR7 access to endosomes.
TLR8 may therefore act as an endogenous brake on TLR7 hyperactivation by competing for endosomal localization. R
RNA recognition in autoimmunity: RNA recognition by human TLR8 in myeloid cells can lead to autoimmune inflammation distinct from the pDC/TLR7 pathway, producing an inflammatory cytokine profile (TNF-α, IL-12) rather than the type I IFN signature of TLR7 activation. R
Bacterial RNA sensing: TLR8 can also detect degradation products of bacterial ribosomal RNA and mRNA, providing an endosomal backup mechanism for detecting bacteria that have been phagocytosed and whose RNA has been released into the lysosomal compartment.
TLR9: The CpG DNA Detector
Location: Endosomal
Dimerization: Homodimer
Signaling: MyD88 (NF-κB and pro-inflammatory cytokines in most cells; massive IFN-α via IRF7 in pDCs)
What TLR9 recognizes:
TLR9 detects unmethylated CpG dinucleotide motifs in single-stranded DNA (ssDNA), which are highly enriched in bacterial and viral genomes compared with mammalian DNA. R
Mammalian DNA contains CpG dinucleotides but they are typically methylated (on the cytosine) and are present at lower frequency due to CpG suppression over evolutionary time.
This difference in CpG methylation and density is the molecular basis by which TLR9 discriminates bacterial/viral DNA from self-DNA under normal conditions.
TLR9 also recognizes:
- Synthetic CpG oligodeoxynucleotides (CpG ODNs): used as vaccine adjuvants and research tools
- DNA from herpes viruses (HSV-1, HSV-2, CMV) and adenovirus in dendritic cells
- DNA:RNA hybrids
- HMGB1-DNA complexes (endogenous alarmin complexes that bypass the methylation barrier in autoimmunity)
- Hemozoin (malaria pigment from Plasmodium falciparum) R
Processing requirement:
TLR9 must be proteolytically cleaved in the endosomal lumen to reach its functional form.
The N-terminal ectodomain fragment produced by cathepsin cleavage remains associated with the C-terminal signaling fragment, and the intact complex is the functional CpG sensor. R
Primary cells and tissues:
In humans, TLR9 is expressed primarily in B cells and plasmacytoid dendritic cells (pDCs), with lower expression in some activated monocyte subsets.
This is a critical species difference from mice: murine TLR9 is expressed much more broadly (monocytes, macrophages, myeloid DCs), which means rodent CpG ODN studies often overpredict inflammatory cytokine responses in humans. R
Disease relevance:
SLE: TLR9 is the primary driver of anti-dsDNA autoantibody production in lupus.
The mechanism: dying cells release chromatin (DNA + protein complexes).
In lupus-prone individuals, autoreactive B cells with BCRs specific for chromatin components (anti-dsDNA B cells) bind and internalize these complexes.
The internalized DNA is delivered to TLR9 in the endosome, providing a co-stimulatory signal that drives B cell activation, differentiation to plasma cells, and anti-dsDNA antibody production without T cell help. R R
Paradoxically, TLR9 deficiency in lupus-prone mice worsens disease in some models (by increasing anti-Sm antibodies and pDC type I IFN production driven by TLR7, which is no longer competed by TLR9 for UNC93B1 access).
This means TLR9 has context-dependent protective and pathogenic roles in lupus. R
CpG ODNs as vaccine adjuvants:
Three classes of synthetic CpG ODNs (A-class: IFN-α induction; B-class: B cell activation; C-class: both) are in clinical development as vaccine adjuvants.
CpG ODN adjuvants markedly enhance the speed and magnitude of antibody and cellular immune responses to vaccines, are well tolerated in humans (unlike rodents where systemic TNF-α storms can occur), and are approved in the hepatitis B vaccine Heplisav-B. R
Cancer:
TLR9 expression in solid tumors is highly context-dependent.
Breast cancer and renal cell carcinoma show inverse relationships between TLR9 expression and prognosis (lower expression correlates with worse outcomes), while prostate cancer, glioma, and non-small cell lung cancer show higher TLR9 expression associated with increased invasion and poor prognosis.
TLR10: The Orphan Receptor With Anti-Inflammatory Properties
Location: Cell surface (and possibly endosomal)
Dimerization: Homodimer, or heterodimer with TLR1 or TLR2
Signaling: MyD88-dependent (but with suppressive rather than pro-inflammatory output; the only TLR known to be predominantly anti-inflammatory)
What TLR10 recognizes:
TLR10 is the least understood of the ten human TLRs and is often called an orphan receptor due to incomplete characterization of its natural ligands. R
Proposed ligands include:
- Listeria monocytogenes components (TLR10 collaborates with TLR2 for Listeria recognition)
- Influenza A virus components
- HIV-1 gp41 protein (TLR10 senses the HIV envelope protein)
- Double-stranded RNA at acidic pH (suggesting possible endosomal localization alongside its cell surface expression) R
Why TLR10 is unique:
TLR10 is the only human TLR that appears to be predominantly anti-inflammatory rather than pro-inflammatory.
TLR10 engagement suppresses pro-inflammatory cytokine production (TNF-α, IL-6, IL-8, IL-12) and may upregulate IL-10 and other anti-inflammatory mediators. R
One proposed mechanism: when TLR10 binds dsRNA in the endosomal compartment, it activates MyD88 but suppresses IRF7-dependent type I IFN expression, simultaneously sequestering dsRNA from TLR3 (which would otherwise produce a pro-inflammatory type I IFN response).
This makes TLR10 a potential endogenous dampener of excessive antiviral innate immune responses. R
Species note:
Mouse TLR10 is non-functional due to retroviral insertion, making it impossible to study TLR10 in standard mouse models.
This is why TLR10 research has lagged far behind other TLRs and why its biology remains incompletely understood.
Primary cells and tissues:
TLR10 is expressed in B cells (highly), pDCs, and monocytes.
Expression on B cells increases with activation and maturation, suggesting a role in regulating B cell inflammatory responses.
TLR10, TLR1, and TLR6 are co-located on chromosome 4p14, and variation in this locus is the major genetic determinant of interindividual differences in TLR1/2-mediated inflammatory responses. R
Disease relevance:
TLR10 polymorphisms are associated with susceptibility to bacterial infections, autoimmune diseases, and certain cancers.
Its anti-inflammatory properties make it a candidate therapeutic target: TLR10 agonists could potentially suppress excessive inflammatory responses in autoimmunity or cytokine storm syndromes without broadly immunosuppressing the host.
Research into TLR10 has accelerated since the COVID-19 pandemic raised interest in mechanisms of excessive innate immune activation. R
TLRs And Autoimmunity: When Self-Recognition Goes Wrong
The endosomal nucleic acid-sensing TLRs (TLR3, TLR7, TLR8, TLR9) evolved to detect viral and bacterial nucleic acids in endosomes after pathogen phagocytosis.
The compartmentalization that normally prevents self-nucleic acids from reaching these sensors can fail in disease.
The self-nucleic acid problem:
In autoimmune conditions, three mechanisms allow self-nucleic acids to access endosomal TLRs:
First, inadequate clearance of apoptotic and necrotic cell debris leaves nucleic acid-containing cellular material available in circulation and tissues.
Patients with SLE have impaired DNase I-dependent clearance of extracellular DNA, and loss-of-function DNase I mutations are directly associated with SLE. R
Second, immune complex-mediated delivery bypasses the endosomal compartmentalization barrier.
Autoantibodies against nuclear antigens bind nuclear material, forming immune complexes that are internalized via Fcγ receptors on macrophages and dendritic cells or via the BCR on autoreactive B cells.
The resulting intracellular delivery of DNA or RNA directly accesses TLR9 or TLR7. R
Third, cationic proteins as carriers: self-DNA in SLE can complex with antimicrobial peptides (such as LL-37/cathelicidin) that convert it into a TLR9-accessible form and protect it from nuclease degradation.
LL-37-DNA complexes can activate pDCs via TLR9 without requiring BCR-mediated internalization.
TLR7 vs TLR9 in SLE:
TLR7 promotes the SLE inflammatory response by driving RNA autoantibody production and massive IFN-α output from pDCs.
TLR9 generates anti-DNA autoantibodies but paradoxically restrains the TLR7-driven IFN response by competing for UNC93B1.
This opposition means that TLR9 deficiency paradoxically worsens some aspects of lupus while improving others. R
TLRs As Therapeutic Targets
Both TLR agonists and antagonists are in clinical development across a range of conditions.
TLR agonists (activating the innate immune response):
- Imiquimod (TLR7/8 agonist): Approved topically for basal cell carcinoma, genital warts, and actinic keratosis. Also studied as a vaccine adjuvant and in cancer immunotherapy.
- CpG ODNs (TLR9 agonist): Used as vaccine adjuvants; approved in Heplisav-B hepatitis B vaccine. In development for allergy immunotherapy, cancer, and anti-infective applications.
- Poly(I:C) / poly-ICLC (TLR3 agonist): Synthetic dsRNA analogs in development as vaccine adjuvants and cancer immunotherapy agents, particularly for anti-tumor T cell responses requiring type I IFN.
- Flagellin-based adjuvants (TLR5 agonist): In development for influenza, pneumococcal, and other vaccines, with particular utility in elderly populations with impaired vaccine responses.
- Resiquimod (TLR7/8 agonist): In development for cancer immunotherapy, acting through stimulation of tumor-associated macrophages and NK cells. R
TLR antagonists (blocking excessive TLR activation):
- Low dose naltrexone (LDN): Acts as a TLR4 antagonist on microglia, specifically blocking the TRIF-IRF3 arm, reducing neuroinflammation in fibromyalgia, Crohn's disease, multiple sclerosis, and chronic pain. R
- (+)-Naltrexone: The non-opioid-receptor-binding enantiomer that retains TLR4 antagonist activity; under investigation as a pure anti-neuroinflammatory without opioid system effects.
- Hydroxychloroquine (TLR7/TLR9 antagonist): An existing antimalarial that blocks endosomal acidification, preventing TLR7 and TLR9 activation by self-nucleic acids. Used for decades in SLE and rheumatoid arthritis without full mechanistic understanding; the TLR7/9 mechanism explains much of its anti-inflammatory benefit. R
- Inhibitory oligonucleotides (TLR7/8/9 antagonists): Synthetic oligonucleotides containing inhibitory sequences that competitively block CpG recognition by TLR9 and ssRNA recognition by TLR7/8. In clinical trials for lupus and other autoimmune conditions.
Natural TLR Modulators {#naturals}
A growing body of preclinical research demonstrates that a range of dietary phytochemicals, fatty acids, and nutrients modulate TLR signaling, particularly TLR4, TLR2, and the endosomal nucleic acid-sensing receptors.
Most evidence is from cell culture and animal models; human clinical data confirming TLR-specific effects at achievable oral doses remain limited for most compounds.
That said, the mechanistic specificity in many studies is strong enough to justify inclusion here as a reference map for researchers and practitioners.
The most consistent theme across virtually all natural TLR modulators: they converge on TLR4 inhibition via MyD88/NF-κB suppression.
TLR2 and the endosomal receptors (TLR7/8/9) are secondary targets for several compounds.
Bioavailability is a persistent caveat for all polyphenols; effects observed at supraphysiological in vitro concentrations may not fully replicate with standard oral dosing.
Berberine (from Coptis chinensis, barberry, Oregon grape)
Primary TLR target: TLR4
Berberine (BBR) is a benzylisoquinoline alkaloid with some of the most mechanistically specific TLR4 data in the natural compound literature.
Molecular docking studies show BBR binds directly to the TLR4 ectodomain, and co-immunoprecipitation experiments in LPS-stimulated macrophages confirm that BBR physically disrupts the TLR4-MyD88 protein-protein interaction. R
This is a distinct upstream mechanism: most natural compounds downregulate TLR4 gene expression or suppress downstream NF-κB; berberine interferes with the TLR4-to-MyD88 signaling handoff at the receptor level. R
In LPS-stimulated macrophages, BBR reduces TLR4 protein expression and suppresses M1 macrophage polarization, reducing TNF-α, IL-6, IL-1β, and MCP-1.
Transcriptional changes in TLR1, TLR2, TLR5, and TLR6 were not the primary mechanism in these experiments, confirming TLR4 specificity. R
In the context of obesity and metabolic inflammation, BBR suppresses adipose tissue macrophage activation via TLR4/MyD88/NF-κB and MAPK pathways, and BBR's metabolite oxyberberine further acts on the TLR4-MyD88-NF-κB pathway in the gut epithelium. R
BBR also suppresses NLRP3 inflammasome activation downstream of TLR4 signaling, relevant to metabolic disease and neuropathic pain. R
Bioavailability note: Standard berberine has poor oral bioavailability (~5%). Dihydroberberine and berberine phytosome formulations significantly improve absorption. R
Black Seed Oil / Thymoquinone (from Nigella sativa)
Primary TLR target: TLR4
Thymoquinone (TQ) is the dominant active constituent of black seed (Nigella sativa) essential oil, accounting for most of its pharmacological effects.
TQ attenuates TLR4 protein and mRNA expression and reduces downstream TLR4-driven inflammatory signaling in multiple models. R
In a liver fibrosis model, TQ reduced TLR4 expression along with pro-inflammatory cytokines, α-SMA, and collagen-I, with concurrent modulation of PI3K/AMPK signaling. R
TQ also potently inhibits NF-κB activation, which is the primary transcriptional output of TLR4/MyD88 signaling. R
An additional mechanism is Nrf2 pathway activation: TQ upregulates heme oxygenase-1 (HO-1) via Nrf2, shifting the cellular redox environment in a way that reduces ROS-driven TLR4 amplification. R
Practical note: Commercial black seed oils vary substantially in TQ content.
Studies screening commercial products found significant variability; look for oils with standardized TQ content or cold-pressed products from quality-controlled sources. R
TQ has cytochrome P450 inhibitory activity (CYP3A4, CYP2D6) that may affect drug metabolism with co-administered medications. R
Curcumin (from Curcuma longa, turmeric)
Primary TLR targets: TLR2, TLR4, TLR9
Curcumin is the most extensively studied natural TLR modulator and acts on multiple levels of TLR4 signaling.
In microglia and macrophages, curcumin reduces TLR4 protein expression and suppresses both arms of TLR4 signaling: the MyD88-NF-κB arm and the TRIF-IRF3 arm. R
In traumatic brain injury models, curcumin significantly reduced TLR4-positive microglial activation, TLR4 protein levels, MyD88, and NF-κB.
TLR4 knockout mice showed similar neuroprotection to curcumin-treated wild-type mice, mechanistically implicating TLR4 inhibition as the primary pathway. R
In a concanavalin A-induced liver injury model, curcumin inhibited TLR2, TLR4, and TLR9 expression simultaneously. R
In autoimmune T lymphocytes, curcumin modulated TLR4 and TLR9 expression, relevant to conditions like rheumatoid arthritis and SLE where autoreactive T cells overexpress these receptors. R
Curcumin also suppresses the NLRP3 inflammasome via TLR4/MyD88/NF-κB, reducing IL-1β production in addition to the upstream TLR4 effects. R
Bioavailability caveat: Standard curcumin powder has notoriously poor oral bioavailability due to rapid conjugation and elimination.
Phytosome (curcumin-phosphatidylcholine complex), nanoparticle, and micellar formulations achieve 20 to 46 times higher bioavailability than standard curcumin. Phytosome curcumin
Piperine (from black pepper) at 20 mg co-administered with 2000 mg standard curcumin increases bioavailability by approximately 20-fold in humans.
DHA and EPA / Omega-3 Fatty Acids (from fish oil, algae oil)
Primary TLR targets: TLR2, TLR4 (primary); TLR3, TLR5, TLR9 (secondary)
Docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) inhibit TLR4 and TLR2 activation by a membrane-level mechanism distinct from all other natural compounds on this list.
Saturated fatty acids (SFAs) activate TLR4 by incorporating into the plasma membrane and promoting TLR4 dimerization and translocation into lipid rafts where downstream signaling molecules are concentrated.
DHA and EPA counter this by displacing SFAs from membrane phospholipids, reducing TLR4 and TLR2 dimerization, and preventing their lipid raft translocation. R
DHA also more broadly inhibits cellular responses to TLR2, TLR3, TLR5, and TLR9 agonists in macrophages, making it the broadest-spectrum natural TLR modulator identified to date. R
TLR4 knockout mice are protected from high-saturated-fat-diet-induced inflammation and insulin resistance, and omega-3 fatty acid supplementation mimics this protection partly by reducing TLR4-driven metabolic inflammation. R
EPA and DHA additionally generate specialized pro-resolving mediators (SPMs) including resolvins, protectins, and maresins that actively resolve inflammation via GPCRs, working downstream of TLR activation rather than at the TLR itself.
This dual mechanism (upstream TLR inhibition via membrane composition changes + downstream resolution via SPMs) makes omega-3 fatty acids uniquely positioned as both preventive and resolving agents.
Practical note: Algae oil provides DHA without the oxidation risk of fish oil; triglyceride-form fish oil has better bioavailability than ethyl ester form. Omega-3 fish oil
EGCG / Epigallocatechin-3-Gallate (from green tea)
Primary TLR target: TLR4
EGCG is the most abundant and pharmacologically active catechin in green tea and has a well-characterized TLR4 inhibition mechanism via a cell-surface receptor: the 67-kDa laminin receptor (67LR).
EGCG binds 67LR on dendritic cells and macrophages, which activates an inhibitory signaling cascade that suppresses TLR4-mediated NF-κB activation and MAPK (ERK1/2, p38, JNK) phosphorylation. R
Through 67LR, EGCG also upregulates Tollip (Toll-interacting protein), an endogenous negative regulator of TLR signaling that prevents IRAK-1 and IRAK-4 from engaging the TLR4 signaling complex. R
In LPS-stimulated dendritic cells, EGCG inhibited expression of CD80, CD86, and MHC class I/II (all required for antigen presentation), along with TNF-α, IL-1β, and IL-6 production, via the 67LR pathway.
In the gut, EGCG reduces TLR4/NF-κB-driven hepatic inflammation in NAFLD models partly by improving gut microbiota composition, increasing short-chain fatty acid producers, and reducing LPS translocation across the gut barrier.
This indirect mechanism (improved gut barrier leading to less LPS exposure and therefore less TLR4 activation) complements the direct 67LR-mediated TLR4 inhibition. R
Sulforaphane, discussed below, directly suppresses TLR4 oligomerization; combining EGCG and sulforaphane produces additive anti-inflammatory effects relevant to the TLR4 axis.
Practical note: EGCG is most bioavailable when consumed as green tea on an empty stomach. Milk proteins bind polyphenols and reduce absorption. Supplement forms standardized to 45 to 50% EGCG are widely available.
Quercetin (from onions, capers, apples, berries)
Primary TLR targets: TLR2, TLR4, TLR9
Quercetin consistently downregulates TLR2, TLR4, and TLR9-dependent MyD88/NF-κB/MAPK signaling across diverse cell types and inflammatory models. R
In macrophages, quercetin reduces TLR4 surface expression, inhibits NF-κB nuclear translocation, and lowers TNF-α, IL-6, and IL-1β production in response to LPS. R
Quercetin's anti-inflammatory effects in inflammatory bowel disease models are partly mediated through inhibition of TLR4 and NF-κB signaling in gut epithelial cells. R
Quercetin also activates Nrf2, upregulating antioxidant enzymes (SOD, catalase, HO-1) that reduce ROS-driven TLR4 amplification.
Because TLR4 signaling produces ROS and ROS in turn amplify TLR4 signaling in a feed-forward loop, Nrf2 activation provides a downstream brake on TLR4-driven inflammation. R
Bioavailability note: Quercetin is poorly absorbed in its aglycone form; quercetin glycoside forms (quercetin-3-glucoside, as found in onions) are better absorbed.
Quercetin phytosome or quercetin with bromelain significantly improves bioavailability in supplement form. Quercetin with bromelain
Resveratrol (from red grapes, Japanese knotweed, berries)
Primary TLR targets: TLR2, TLR4, TLR9
Resveratrol downregulates TLR2, TLR4, and TLR9-dependent MyD88/NF-κB/MAPK pathways and elevates IL-10 in multiple inflammatory models. R
In ulcerative colitis models, resveratrol attenuates TLR4-driven gut inflammation and restores epithelial barrier function through NF-κB suppression. R
Resveratrol additionally induces the cathelicidin antimicrobial peptide (CAMP) gene via a VDR-independent mechanism, and synergizes with vitamin D to further amplify CAMP gene expression via stilbenoid-specific pathways. R
Because cathelicidin (LL-37) is a key effector of TLR-activated innate immunity, this CAMP-inducing property positions resveratrol as both a TLR inhibitor (suppressing excessive TLR signaling) and an innate immune enhancer (boosting the downstream effector response).
Resveratrol also activates SIRT1, which deacetylates and deactivates NF-κB p65, providing a post-translational brake on TLR4-driven inflammatory gene transcription.
Bioavailability note: Trans-resveratrol is rapidly conjugated after oral absorption; peak plasma concentrations from food sources are very low.
Resveratrol with piperine or in micronized formulations achieves meaningfully higher bioavailability.
Pterostilbene, a methylated analog from blueberries, has greater bioavailability and similar or stronger TLR-modulating activity. Trans-resveratrol
Sulforaphane (from broccoli sprouts, crucifers)
Primary TLR target: TLR4
Sulforaphane has a structurally unique TLR4 mechanism: it directly suppresses TLR4 oligomerization via thiol-dependent alkylation.
TLR4 activation requires dimerization; sulforaphane's isothiocyanate group forms covalent bonds with cysteine residues at the TLR4 dimer interface, preventing the conformational changes required for dimerization and downstream signaling. R
This thiol-dependent mechanism is distinct from the gene-expression-level suppression seen with most other natural compounds and means sulforaphane can act rapidly (minutes) rather than requiring hours for protein expression changes.
Sulforaphane is also the most potent natural Nrf2 activator known.
By inhibiting Keap1 (the Nrf2 repressor) through cysteine alkylation, sulforaphane drives robust upregulation of HO-1, NQO1, glutathione synthesis genes, and other antioxidant response elements.
This Nrf2 activity breaks the ROS-TLR4 amplification loop that sustains chronic neuroinflammation and metabolic inflammation. R
Sulforaphane reduces NLRP3 inflammasome activation downstream of TLR4, lowering IL-1β and caspase-1 cleavage.
In the gut, sulforaphane supports tight junction integrity and reduces LPS translocation, providing the same indirect TLR4 benefit as EGCG. R
Practical note: Broccoli sprouts contain 10 to 100 times more glucoraphanin (the sulforaphane precursor) than mature broccoli.
Sulforaphane is generated by myrosinase enzyme action on glucoraphanin; heat destroys myrosinase, so raw sprouts or supplements containing both glucoraphanin and myrosinase (active sulforaphane) are preferred over cooked. Sulforaphane / broccoli sprout extract
Vitamin D3 / Calcitriol (from sunlight, fatty fish, fortified foods)
Primary TLR targets: TLR2, TLR4, TLR9 (expression downregulation); also modulates TLR2-triggered innate responses via cathelicidin induction
Vitamin D has a dual and sometimes paradoxical role in TLR biology: it downregulates TLR expression while simultaneously enhancing the antimicrobial effector responses triggered by those same TLRs.
The active form of vitamin D (1,25-dihydroxyvitamin D3, or calcitriol) decreases the expression of TLR2, TLR4, and TLR9 in monocytes and macrophages, reducing their sensitivity to pathogen-derived PAMPs and endogenous DAMPs. R
Calcitriol also inhibits LPS-induced cytokine production (IL-6, IL-23, TNF-α, IL-1β) through activation of MAPK phosphatase-1 (MKP-1), which dephosphorylates and inactivates MAPK signaling downstream of TLR4. R
At the transcriptional level, calcitriol induces IκB (the NF-κB inhibitor), preventing NF-κB nuclear translocation in response to TLR4 stimulation. R
The enhancement side: when TLR2/1 detects a mycobacterial ligand, it triggers local upregulation of the 1-α-hydroxylase enzyme (CYP27B1) in macrophages, which converts circulating 25(OH)D3 to active calcitriol on-site.
This locally generated calcitriol then drives cathelicidin (LL-37) production via VDR binding to the cathelicidin gene's VDRE.
LL-37 then directly kills intracellular bacteria and facilitates autophagosome formation for pathogen clearance. R
This means TLR2 activation in vitamin-D-sufficient individuals triggers a self-reinforcing antibacterial cycle; in vitamin-D-deficient individuals, this antimicrobial loop is broken.
Butyrate (from dietary fiber fermentation) synergizes with vitamin D by increasing histone acetylation at the cathelicidin gene promoter, amplifying vitamin D-induced CAMP expression independent of VDR occupancy. R
Resveratrol also synergizes with calcitriol to increase CAMP expression through a separate SIRT1/kinase pathway. R
Practical note: The TLR-suppressive effects of vitamin D require sufficient serum 25(OH)D3 levels; most immune effects are apparent above 40 to 60 ng/mL.
Vitamin D testing is available through the lab store (Foundation Zoomer panel includes vitamin D).
Baicalin / Baicalein (from Scutellaria baicalensis, Chinese skullcap)
Primary TLR targets: TLR2, TLR4, TLR9
Baicalin (the glycoside form) and baicalein (the aglycone) consistently downregulate TLR2, TLR4, and TLR9-dependent MyD88/NF-κB/MAPK signaling in multiple inflammatory models. R
Baicalin reduces pro-inflammatory cytokine production (IL-1β, IL-6, TNF-α) and elevates IL-10, matching the anti-inflammatory profile of curcumin and quercetin on the same TLR targets.
Baicalin activates Nrf2 and upregulates HO-1, providing additional ROS suppression that reduces TLR4 amplification.
It is among the most well-studied Chinese herbal medicine compounds for innate immune modulation and has a longer clinical use history than many other compounds on this list.
Glycyrrhizin / Glycyrrhizinic Acid (from licorice root)
Primary TLR targets: TLR2, TLR4, TLR9
Glycyrrhizin and its active metabolite 18β-glycyrrhetinic acid downregulate TLR2, TLR4, and TLR9-dependent MyD88/NF-κB/MAPK pathways. R
A key additional mechanism is inhibition of HMGB1 (high mobility group box 1), a major endogenous TLR2 and TLR4 DAMP released during cell death.
Glycyrrhizin directly binds HMGB1 and blocks its interaction with TLR2 and TLR4, reducing sterile inflammation driven by damaged tissue. R
This HMGB1-blocking property distinguishes glycyrrhizin from most other natural TLR modulators, which act on the receptor or downstream signaling rather than on an endogenous ligand.
Important caveat: High-dose or chronic glycyrrhizin consumption (>100 mg/day glycyrrhizic acid) can cause pseudohyperaldosteronism (sodium retention, hypokalemia, hypertension) via inhibition of 11β-hydroxysteroid dehydrogenase.
Deglycyrrhizinated licorice (DGL) removes glycyrrhizic acid for gut-directed use, but also loses the TLR-modulating glycyrrhizin fraction.
Therapeutic TLR-modulating doses require intact glycyrrhizin.
Butyrate and Short-Chain Fatty Acids (from dietary fiber fermentation)
Primary TLR targets: TLR4 (expression and signaling suppression); also amplifies TLR-triggered innate responses via cathelicidin
Butyrate, the principal short-chain fatty acid (SCFA) produced by gut microbial fermentation of dietary fiber, suppresses TLR4/NF-κB-driven inflammation in intestinal epithelial cells and macrophages through two distinct mechanisms.
First, butyrate directly inhibits histone deacetylases (HDACs), particularly HDAC1, HDAC2, and HDAC3, which reprograms macrophage gene expression toward an anti-inflammatory state.
HDAC inhibition by butyrate reduces TLR4-triggered NF-κB target gene transcription while upregulating anti-inflammatory genes including IL-10. R
Second, butyrate strengthens tight junction proteins (claudin-1, occludin, ZO-1) in the gut epithelium, physically reducing the amount of LPS that translocates from the gut lumen into portal circulation.
Less circulating LPS means less TLR4 stimulation on peripheral macrophages, adipose tissue immune cells, and hepatic Kupffer cells, reducing systemic inflammatory tone. R
The butyrate-vitamin D synergy for cathelicidin induction is directly relevant to TLR2-triggered antimicrobial responses: butyrate increases histone acetylation at the cathelicidin promoter, allowing vitamin D-bound VDR to drive stronger CAMP transcription than vitamin D alone. R
Butyrate is produced endogenously from resistant starch and other fermentable fibers by butyrate-producing bacteria (primarily Faecalibacterium prausnitzii, Roseburia intestinalis, Eubacterium rectale).
Dietary resistant starch, psyllium, inulin, and arabinoxylan all increase butyrate production.
Direct butyrate supplementation (sodium butyrate, tributyrin) is also available but largely acts in the colon rather than systemically.
Beta-Caryophyllene (from cannabis, black pepper, cloves, hops)
Primary TLR targets: TLR4 (via CD14/TLR4/MD-2 complex inhibition through CB2R)
Beta-caryophyllene is a dietary sesquiterpene terpene and the only known dietary compound that is both a TLR4 pathway inhibitor and a selective CB2 receptor agonist.
Via CB2R activation, beta-caryophyllene inhibits the CD14/TLR4/MD-2 signaling complex, reducing IL-1β, IL-6, IL-8, and TNF-α production. R
This places beta-caryophyllene at the intersection of the cannabinoid and TLR4 systems, making it uniquely relevant in conditions where both TLR4-driven neuroinflammation and endocannabinoid tone are therapeutic targets (chronic pain, neuroinflammation, post-COVID).
Beta-caryophyllene is an FDA-approved food additive (GRAS status), is found in black pepper, cloves, hops, and cannabis at reasonable dietary amounts, and lacks psychoactivity.
Full-spectrum cannabis preparations that contain beta-caryophyllene alongside CBD produce additive TLR4-suppressive effects.
Summary Table: Natural TLR Modulators
| Compound | Primary TLR Targets | Key Mechanism | Bioavailability Caveat |
|---|---|---|---|
| Berberine | TLR4 | Disrupts TLR4-MyD88 protein interaction; direct TLR4 binding | Poor (~5%); use phytosome or dihydroberberine |
| Black seed oil (TQ) | TLR4 | Reduces TLR4 expression; Nrf2/HO-1 induction; NF-κB inhibition | Variable TQ content between products |
| Butyrate / SCFAs | TLR4 | HDAC inhibition; gut barrier repair reducing LPS translocation; cathelicidin synergy with vitamin D | Colonic production depends on fiber intake |
| Curcumin | TLR2, TLR4, TLR9 | Reduces TLR4/MyD88/NF-κB/IRF3 protein expression; NLRP3 suppression | Very poor standard form; use phytosome/micellar/nanoparticle |
| EGCG (green tea) | TLR4 | 67LR-mediated TLR4 inhibition; Tollip upregulation; gut barrier support | Better on empty stomach; milk proteins reduce absorption |
| Omega-3 EPA/DHA | TLR2, TLR4 (primary); TLR3, TLR5, TLR9 | Membrane-level: prevents TLR4/TLR2 dimerization and lipid raft translocation; generates SPMs | TG form better than EE; algae oil avoids oxidation risk |
| Quercetin | TLR2, TLR4, TLR9 | Reduces TLR4 expression; NF-κB inhibition; Nrf2/HO-1 induction | Poor aglycone; glucoside or phytosome forms preferred |
| Resveratrol | TLR2, TLR4, TLR9 | TLR4/NF-κB/MyD88 downregulation; SIRT1-mediated NF-κB deacetylation; CAMP gene induction | Very poor; micronized or piperine co-administration needed |
| Sulforaphane | TLR4 | Direct TLR4 oligomerization inhibition via thiol alkylation; Nrf2 activation | Cook destroys myrosinase; use raw sprouts or active-sulforaphane supplements |
| Vitamin D3 | TLR2, TLR4, TLR9 | Reduces TLR expression; MKP-1 activation; IκB induction; TLR2-triggered cathelicidin loop | Requires serum 25(OH)D above 40 ng/mL; test before and during supplementation |
| Baicalin | TLR2, TLR4, TLR9 | MyD88/NF-κB/MAPK suppression; Nrf2/HO-1 induction; IL-10 elevation | Moderate; aglycone baicalein may have better membrane penetration |
| Beta-caryophyllene | TLR4 | CB2R agonism inhibits CD14/TLR4/MD-2 complex; non-psychoactive | Good; GRAS dietary compound |
| Glycyrrhizin | TLR2, TLR4, TLR9 | MyD88/NF-κB/MAPK suppression; direct HMGB1 blockade preventing DAMP-driven TLR activation | Pseudohyperaldosteronism risk at high/chronic doses |
Mechanisms Of Action
Simple:
- Human cells express ten TLRs (TLR1 through TLR10), each positioned to detect specific molecular signatures of bacteria, viruses, fungi, or parasites.
- Cell surface TLRs (TLR1, TLR2, TLR4, TLR5, TLR6, TLR10) detect bacterial membrane components: lipids, lipoproteins, and flagellin.
- Endosomal TLRs (TLR3, TLR7, TLR8, TLR9) detect microbial nucleic acids after phagocytosis: dsRNA (TLR3), ssRNA (TLR7, TLR8), and unmethylated CpG DNA (TLR9).
- All TLRs except TLR3 signal through MyD88 to activate NF-κB and produce pro-inflammatory cytokines; TLR3 (and TLR4 in its endosomal phase) uses TRIF to activate IRF3 and produce type I interferons.
- TLR2 functions only as a heterodimer: with TLR1 for triacyl lipopeptides, with TLR6 for diacyl lipopeptides.
- TLR4 is the most complex TLR, requiring co-receptors CD14 and MD-2, and using all four TIR adapters (TIRAP, MyD88, TRAM, TRIF) across two activation phases.
- TLR9 drives anti-DNA autoantibody production in lupus when self-DNA is delivered to endosomal B cells via immune complexes; TLR7 drives the anti-RNA autoantibody response and massive IFN-α from pDCs.
- TLR10 is the only anti-inflammatory TLR, suppressing rather than promoting cytokine production; it is non-functional in mice, limiting research.
- Hydroxychloroquine works in lupus partly by blocking endosomal acidification, preventing TLR7 and TLR9 activation by self-nucleic acids.
- LDN targets the TLR4 TRIF-IRF3 axis on microglia, reducing chronic neuroinflammation.
Advanced:
- Ligand discrimination at TLR2 heterodimers: The structural basis for TLR2/TLR1 vs TLR2/TLR6 specificity lies in the hydrophobic channel within the TLR1 or TLR6 ectodomain. TLR1's channel (formed by residues that maintain an open hydrophobic pocket) accommodates the amide-linked third acyl chain of triacylated lipopeptides. TLR6's equivalent channel is occluded by phenylalanine 343 and leucine 344, physically excluding the third acyl chain and restricting TLR6 to diacylated ligands. Crystal structures of TLR1/TLR2-Pam3CSK4 and TLR2/TLR6-Pam2CSK4 complexes confirm this mechanism. R
- TLR4 dual-phase signaling and TRIF pathway compartmentalization: TLR4 initiates MyD88-dependent NF-κB at the plasma membrane (early phase), then undergoes clathrin-mediated endocytosis. In the endosome, TRAM recruits TRIF independently of TIRAP/MyD88. TRIF assembles a signalosome containing TRAF3, which recruits TBK1 via TRAF3-TBK1 interaction. TBK1 and IKKε then phosphorylate IRF3 at serine 396, inducing IRF3 dimerization, cytoplasmic release from its inhibitor, nuclear translocation, and binding to the positive regulatory domain (PRD) of the IFN-β promoter. The compartmentalization of TRIF signaling to endosomes explains why TLR4 but not TLR5 (which also signals from the surface) produces type I interferon: TLR4 undergoes ligand-induced endocytosis while TLR5 does not. R
- UNC93B1 competition and TLR7/TLR9 balance in SLE: UNC93B1 is the ER export chaperone required for TLR7, TLR8, and TLR9 to exit the ER and traffic to endosomes. The three receptors compete for binding to UNC93B1. When TLR9 is absent, TLR7 access to endosomes increases because it no longer competes with TLR9 for UNC93B1 occupancy. This explains why TLR9 deficiency paradoxically worsens some lupus manifestations: TLR7 is released from competition, gaining enhanced endosomal access and driving stronger IFN-α and anti-Sm antibody responses. Conversely, TLR9 generates anti-dsDNA antibodies but moderates the IFN-α response. Therapeutically, this means interventions that specifically inhibit TLR9 without affecting TLR7 may worsen the type I IFN signature in lupus, and balanced inhibition of both receptors may be required. R
- TLR7 two-site activation model: TLR7 activation requires two distinct small-molecule binding events that together induce the homodimer's closed, signaling-competent configuration. Site 1 (at the homodimer interface) binds a guanosine-like compound or guanosine from RNA degradation products. Site 2 (adjacent to site 1 on the same protomer) binds a short ssRNA oligonucleotide. Single-site occupancy produces weak or no activation; both sites must be occupied simultaneously. This two-site requirement means TLR7 senses intact RNA (which provides both nucleosides and oligonucleotides) rather than isolated nucleosides or degradation products alone, providing a mechanistic discrimination between pathological RNA and normal metabolic RNA turnover products. R
Genetics
TLR1 I602S (rs4833095, also reported as T1805G):
This missense variant in TLR1 reduces surface expression of TLR1 and blunts lipopeptide-induced cytokine secretion from monocytes.
The variant is associated with susceptibility to tuberculosis and leprosy in multiple human populations. R
TLR2 R753Q (rs5743708):
A missense variant in TLR2 associated with susceptibility to tuberculosis (Vietnamese populations) and other bacterial infections.
The R753Q substitution impairs TLR2 signaling and reduces inflammatory cytokine production in response to TLR2 ligands.
Also associated with susceptibility to septic shock from gram-positive organisms in some populations.
TLR4 Asp299Gly (rs4986790) and Thr399Ile (rs4986791):
Two common co-inheriting variants that reduce LPS responsiveness by impairing TLR4-MD-2 dimerization.
Associated with reduced inflammatory cytokine production in response to LPS, reduced gram-negative sepsis risk, and increased susceptibility to some intracellular infections.
Also associated with earlier onset of atherosclerosis and reduced inflammatory markers in some cardiovascular studies.
TLR5 stop codon (rs5744168, F616L):
A premature stop codon in TLR5 creating a non-functional receptor in approximately 7 to 10% of individuals with European ancestry.
Associated with protection from SLE (by reducing overall inflammatory tone) but increased susceptibility to Legionella pneumophila pneumonia (because TLR5-driven flagellin recognition is non-redundantly required for defense against this organism).
Also associated with reduced vaccine efficacy to flagellin-based vaccines. R
TLR7 gain-of-function variants (including Gln11Leu and others):
Gain-of-function variants in TLR7 increase ssRNA sensing activity and are strongly associated with increased SLE susceptibility and severity, earlier onset, and more aggressive renal disease.
The X-chromosome location of TLR7 and incomplete X-inactivation in some females allows higher TLR7 expression per cell in women, contributing to the 9:1 female-to-male ratio of SLE. R
TLR9 rs352140 (C/T):
The T allele of this TLR9 coding polymorphism is associated with increased anti-dsDNA antibody production, renal disease, and immunologic disorder severity in SLE patients.
TLR9 polymorphisms also affect susceptibility to tuberculosis (along with TLR2, TLR4) and modify the risk of certain cancers. R
UNC93B1 loss-of-function (H412R):
The 3d mutation in UNC93B1 (histidine 412 to arginine) prevents UNC93B1 from trafficking intracellular TLRs (TLR3, TLR7, TLR8, TLR9) from the ER to endosomes.
Human loss-of-function mutations in UNC93B1 cause selective susceptibility to herpes simplex encephalitis by preventing TLR3 from accessing the endosomal compartment where it would otherwise detect HSV-1 nucleic acids and drive IFN production in CNS cells. R
More Research
- Trained immunity and TLRs: Low-dose stimulation of TLR2, TLR4, TLR5, TLR3, and TLR7/8 can epigenetically reprogram monocytes and macrophages into a state of enhanced innate immune responsiveness (trained immunity), where they respond more vigorously to secondary challenges.
High-dose stimulation by the same ligands produces immune tolerance instead.
The same dose-response dichotomy seen with LDN's TLR4 modulation (LDN doses produce tolerance-like microglial modulation; standard anti-infective doses activate the full response) appears to be a general feature of TLR biology. R
TLR9 is an exception: both low and high doses of CpG ODN in human monocytes fail to induce trained immunity, suggesting TLR9 does not participate in this epigenetic reprogramming despite being a strong activator of immediate innate responses.
- Gut microbiome and TLR homeostasis: The composition of the gut microbiota determines the chronic background level of TLR stimulation that shapes innate immune tone systemically.
Gut dysbiosis increases the LPS load crossing a leaky gut wall, chronically stimulating TLR4 on peripheral macrophages and contributing to metabolic inflammation, insulin resistance, and cardiovascular disease.
Resistant starch, dietary fiber, and butyrate-raising interventions reduce gut permeability and LPS translocation, lowering chronic TLR4 tone as a downstream metabolic benefit.
Simultaneously, commensal bacteria provide low-level TLR2 and TLR5 stimulation that is required for normal mucosal immune development and intestinal IgA production. R
- TLRs and cancer: TLR expression is altered in virtually all solid tumors, but in highly tumor-specific and often opposite ways.
TLR agonists in tumors can activate immune cells in the tumor microenvironment to mount anti-tumor responses (the rationale for TLR7/8/9 agonist cancer immunotherapy) or can directly stimulate cancer cell survival and proliferation via NF-κB (the pro-tumor effect).
Which effect dominates depends on the TLR, the cancer type, and the local cytokine environment.
- TLRs and aging (immunosenescence): Aging is associated with increased basal TLR activation (contributing to the chronic low-grade inflammation called "inflammaging") combined with impaired TLR-driven adaptive immune priming after infection or vaccination.
The paradox of inflammaging is that microglia and macrophages are chronically activated by accumulated DAMPs from age-related cell damage, yet respond less efficiently to acute TLR stimulation by pathogens due to tolerance mechanisms.
Flagellin-based TLR5 adjuvants specifically enhance vaccine responses in aged mice and elderly humans, suggesting TLR5 activation may help break the tolerance that impairs vaccine efficacy in the elderly. R
- TLR crosstalk: Individual TLRs do not operate in isolation.
TLR2 and TLR4 cooperate to recognize mycobacterial 38-kDa glycolipid antigen.
TLR4 and TLR8 can bind together to recognize TLR8 ligands from M. tuberculosis.
TLR9 and TLR2 cooperate for optimal resistance to mycobacterial infection in animal models.
This crosstalk means that the functional immune response to a complex pathogen reflects the simultaneous activation of multiple TLRs, and that pharmacological targeting of one TLR will inevitably affect responses mediated by cooperating receptors.
Understanding TLR crosstalk is necessary for rational design of both vaccine adjuvant combinations and anti-inflammatory TLR inhibition strategies.
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Quercetin
500mg 2x/day
Vitamin D3 + K2
5000 IU + 200mcg/day
DAO Enzyme
1 cap before meals