T Helper Cell Subsets: Th1, Th2, Th17, Tregs, Th9, Th22, And Tfh Explained
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.
Every chronic illness, autoimmune condition, allergy, and cancer has an immune context, and that context is largely written by which T helper cell subsets are dominant, which are suppressed, and whether the regulatory system is strong enough to prevent collateral damage.
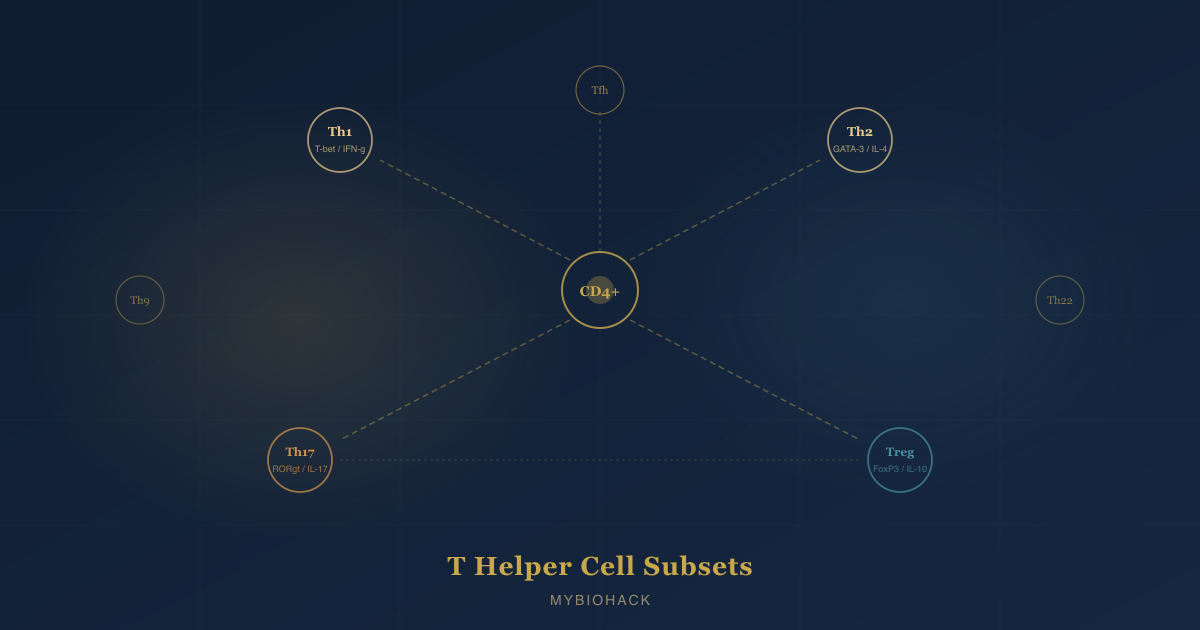
In this post, we will discuss what T helper (Th) cells are, how naive CD4+ T cells decide which subset to become, the full set of known subsets including Th1, Th2, Th17, regulatory T cells (Tregs), Th9, Th22, and T follicular helper (Tfh) cells, what each one does, how they cross-regulate each other, which diseases are associated with each subset's dysregulation, and what the Th17/Treg balance means for chronic disease.
The Basics: What T Helper Cells Are And How Differentiation Works
T helper (Th) cells are CD4+ lymphocytes that orchestrate adaptive immune responses by secreting cytokines that activate and direct other immune cells. R
They do not kill pathogens or infected cells directly (that is the job of CD8+ cytotoxic T cells and natural killer (NK) cells).
Instead, they serve as the immune system's command layer, instructing macrophages, B cells, cytotoxic T cells, neutrophils, mast cells, and eosinophils on what kind of threat is present and what response to mount. R
The differentiation decision:
All Th cells begin as naive CD4+ T cells, which emerge from the thymus uncommitted to any effector fate. R
Three signals converge to determine which subset a naive T cell becomes:
- Signal 1: The T cell receptor (TCR) recognizing a specific peptide antigen presented by major histocompatibility complex class II (MHC-II) on an antigen-presenting cell (APC), most commonly a dendritic cell (DC). R
- Signal 2: Costimulatory receptor engagement, principally CD28 binding B7 ligands (CD80/CD86) on the APC surface. R
- Signal 3: The cytokine milieu produced by the APC and the local tissue environment in response to the nature of the threat. R
Signal 3 is the decisive signal for subset fate.
Different cytokines activate different members of the signal transducer and activator of transcription (STAT) family, and each STAT drives expression of a lineage-defining master transcription factor that locks the cell into a subset identity. R
The cytokine-to-STAT-to-master-transcription-factor logic for each subset is summarized below and expanded in the individual sections.
Plasticity:
Committed Th cell subsets are not permanently fixed.
Under altered cytokine environments, cells can partially or fully convert to another phenotype, a phenomenon called plasticity. R
For example, Th17 cells stimulated by interferon-gamma (IFN-γ) can convert toward Th1, and natural Tregs (nTregs) can convert toward Th17 in the presence of interleukin-6 (IL-6). R
This plasticity is clinically significant because it means that once-suppressed disease can relapse under new inflammatory conditions even if the original triggering antigen is gone.
Th1: The Intracellular Pathogen Fighter
Polarizing cytokines: Interleukin-12 (IL-12) and IFN-γ. R
STAT pathway: IL-12 activates signal transducer and activator of transcription 4 (STAT4); IFN-γ activates STAT1. R
Master transcription factor: T-bet (T-box transcription factor TBX21). R
Signature cytokines produced: IFN-γ and tumor necrosis factor-alpha (TNF-α). R
What Th1 cells do:
Th1 cells coordinate immunity against intracellular pathogens: bacteria that survive inside macrophages (such as Mycobacterium tuberculosis, Listeria, Salmonella), intracellular parasites (such as Leishmania), and viruses. R
IFN-γ produced by Th1 cells activates macrophages into a classical (M1) activation state, dramatically increasing their ability to produce ROS, nitric oxide (NO) via inducible nitric oxide synthase (iNOS), and lysosomal enzymes capable of destroying intracellular pathogens. R
Th1 cells also activate CD8+ cytotoxic T lymphocytes (CTLs) via IFN-γ and CD40L signaling, supporting the killing of virally infected or cancerous cells. R
IFN-γ drives immunoglobulin G2a (IgG2a) isotype switching in B cells, the antibody isotype best suited for opsonization and complement activation against intracellular pathogens. R
Diseases driven by Th1 excess:
A pathologically dominant Th1 response is associated with:
- Crohn's disease (granulomatous inflammation of intestinal wall)
- Type 1 diabetes (T1D) (destruction of pancreatic beta cells)
- Rheumatoid arthritis (RA) (joint synovial inflammation)
- Multiple sclerosis (MS) (early phases, later dominated by Th17)
- Hashimoto's thyroiditis
- Organ transplant rejection
Th1 insufficiency:
Inadequate Th1 function leaves the host vulnerable to intracellular bacterial infections, mycobacterial disease (including tuberculosis), viral infections, and reduces anti-tumor immunity via reduced CTL activation.
Th2: The Parasite And Allergy Arm
Polarizing cytokine: Interleukin-4 (IL-4). R
STAT pathway: IL-4 activates STAT6. R
Master transcription factor: GATA-3 (GATA-binding protein 3). R
Signature cytokines produced: IL-4, interleukin-5 (IL-5), and interleukin-13 (IL-13). R
What Th2 cells do:
Th2 cells coordinate immunity against helminths (parasitic worms) and extracellular parasites. R
IL-4 and IL-13 drive immunoglobulin E (IgE) class-switching in B cells: IgE binds mast cells and basophils, sensitizing them to rapid degranulation upon antigen re-exposure. R
IL-5 activates and recruits eosinophils, which release cytotoxic granule proteins lethal to large parasites that cytotoxic T cells cannot handle.
IL-13 stimulates intestinal epithelial cell turnover, increases intestinal smooth muscle contractility, and drives mucus production, all of which physically expel parasites from the gut. R
Where Th2 goes wrong: allergic disease
In the developed world where parasitic infection is rare, Th2 responses are most often activated not by helminths but by environmental antigens (pollen, dust mites, food proteins, animal dander).
This misdirected Th2 response produces:
- Allergic asthma (IL-4/IL-13 driven airway inflammation and mucus hypersecretion)
- Atopic dermatitis / eczema (IL-4/IL-13 driven skin barrier disruption and IgE sensitization)
- Allergic rhinitis
- Food allergies (IgE-mediated anaphylaxis risk)
- Eosinophilic esophagitis (EoE)
- Systemic lupus erythematosus (SLE) has Th2 involvement (B cell overactivation)
Th2 insufficiency:
Low Th2 capacity impairs the ability to clear helminthic parasites and results in chronic infection.
In the context of Western chronic illness, Th2 insufficiency is rarely the presenting problem: Th2 excess (allergy and atopy) is far more common.
Th17: The Fungal And Bacterial Frontline
Polarizing cytokines: Transforming growth factor-beta (TGF-β) plus IL-6 (or IL-21) for initial differentiation; IL-23 for maintenance and expansion; IL-1β amplifies the response. R
STAT pathway: IL-6 and IL-21 activate STAT3; STAT3 then drives expression of the master transcription factor. R
Master transcription factor: RORγt (retinoic acid receptor-related orphan receptor gamma t), with RORα as a cooperative secondary factor. R
Signature cytokines produced: IL-17A, IL-17F, and IL-22. R
What Th17 cells do:
Th17 cells coordinate immunity against extracellular bacteria and fungi, particularly at epithelial barrier surfaces: the intestinal mucosa, lung epithelium, and skin. R
IL-17A and IL-17F stimulate epithelial cells, fibroblasts, and stromal cells to produce granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage CSF (GM-CSF), which drive neutrophil production and recruitment to sites of infection. R
Neutrophils are the primary killers of extracellular bacteria and fungi, so Th17 activity drives the innate antimicrobial first response.
IL-22 maintains intestinal epithelial barrier integrity, induces antimicrobial peptide production by Paneth cells, and promotes epithelial regeneration after infection or injury. R
IL-23 is critical for Th17 pathogenicity:
Initial Th17 differentiation requires TGF-β plus IL-6, but subsequent exposure to IL-23 (produced by activated macrophages and dendritic cells) drives the cells toward a more pathogenic, self-sustaining phenotype with higher IL-17 production and greater capacity to cause tissue damage. R
This distinction matters therapeutically: drugs targeting IL-23 (risankizumab, guselkumab) have become major tools in inflammatory disease precisely because IL-23 drives the pathogenic arm of Th17 without affecting initial TGF-β/IL-6 differentiation.
Diseases driven by Th17 excess:
Many diseases previously attributed entirely to Th1 cells are now understood to be driven by Th17 cells. R
The evidence: mice deficient in IFN-γ (the Th1 cytokine) were still fully capable of developing severe collagen-induced arthritis and experimental autoimmune encephalomyelitis, conditions previously considered Th1-driven. R
Th17-associated diseases include:
- Psoriasis (strongest Th17 association; IL-17 blockade with secukinumab, ixekizumab is highly effective)
- Inflammatory bowel disease (IBD), particularly Crohn's disease (joint Th1/Th17 involvement)
- Ankylosing spondylitis (axial spondyloarthritis)
- Rheumatoid arthritis
- MS (later pathogenic phase)
- Asthma (neutrophilic, non-eosinophilic variant)
- Candidal overgrowth susceptibility when Th17 is deficient (inherited STAT3 mutations and IL-17 defects produce chronic mucocutaneous candidiasis)
Th17 insufficiency:
Low Th17 capacity leaves mucosal surfaces vulnerable to extracellular bacterial and fungal invasion.
Primary immunodeficiencies affecting the IL-17 axis produce chronic mucocutaneous candidiasis (CMC) as a hallmark feature.
Regulatory T Cells (Tregs): The Immune Brake
Two major origins:
Natural Tregs (nTregs), also called thymic Tregs (tTregs), develop in the thymus from CD4+ thymocytes that recognize self-antigens with moderate affinity. R
They comprise roughly 5 to 10% of peripheral CD4+ T cells and are marked by constitutive expression of CD25 (the IL-2 receptor alpha chain) and the master transcription factor FoxP3. R
Induced Tregs (iTregs), also called peripheral Tregs (pTregs), arise from naive CD4+ conventional T cells in the periphery in response to TGF-β, IL-2, and retinoic acid (RA). R
iTregs include several subpopulations:
- Tr1 cells: FoxP3-negative; defined by high IL-10 production; most abundant at intestinal mucosal surfaces. R
- Th3 cells: Defined by TGF-β secretion; first identified in mucosal tolerance to oral antigens.
- iTr35 cells: IL-35-dependent regulatory cells without constitutive FoxP3 expression. R
Master transcription factor: FoxP3 (forkhead box P3, an X-linked transcription factor). R
Mutations in FoxP3 in humans cause IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked), a catastrophic multi-organ autoimmune disease of infancy, demonstrating that FoxP3+ Tregs are non-redundantly required for immune self-tolerance. R
Polarizing signals for iTregs: TGF-β plus IL-2 (in the absence of pro-inflammatory cytokines). R
STAT pathway: IL-2 activates STAT5, which promotes FoxP3 expression and Treg maintenance. R
How Tregs suppress immunity:
Tregs suppress via multiple non-redundant mechanisms: R
- Inhibitory cytokine secretion: IL-10, TGF-β, and IL-35 suppress effector T cell activation and APC function.
- CTLA-4-mediated co-stimulation blockade: Cytotoxic T lymphocyte antigen-4 (CTLA-4), constitutively expressed by nTregs, competes with CD28 for B7 ligands on APCs, reducing costimulatory signal to effector T cells. CTLA-4 also strips B7 from APC surfaces via trans-endocytosis.
- IL-2 consumption: Tregs express high-affinity IL-2 receptor (CD25) constitutively and consume local IL-2, depriving effector T cells of their growth and survival signal.
- Metabolic disruption: Tregs produce adenosine via CD73 and CD39 enzymes, which suppresses T cell activation through A2A adenosine receptor signaling.
- Direct cytolysis: nTregs can kill effector T cells via granzyme B and perforin in a contact-dependent manner. R
nTreg vs. iTreg stability:
This distinction matters clinically.
nTregs have a demethylated FoxP3 Treg-specific demethylated region (TSDR) in the FoxP3 locus, which confers epigenetic stability of FoxP3 expression even under inflammatory conditions. R
iTregs have a partially methylated TSDR and are intrinsically less stable: under inflammatory conditions (particularly IL-6 and IL-1β exposure), iTregs can lose FoxP3 expression and convert toward Th17 or Th1 phenotypes, potentially amplifying rather than suppressing the response they were meant to control. R
Diseases driven by Treg insufficiency or dysfunction:
- Virtually all autoimmune diseases (insufficient suppression of autoreactive Th1 and Th17 cells)
- Food allergies and atopic disease (insufficient mucosal tolerance via Tr1 cells)
- IBD (insufficient intestinal Treg activity)
- Graft-versus-host disease (GvHD)
- Recurrent pregnancy loss (RPL) (insufficient tolerance at maternal-fetal interface)
- IPEX syndrome (FoxP3 mutation, catastrophic multi-organ autoimmunity)
Diseases driven by Treg excess:
- Cancer: Tumors recruit and expand Tregs in the tumor microenvironment (TME), using TGF-β to induce iTregs and chemokines to recruit nTregs, suppressing anti-tumor CTL activity. Treg accumulation in solid tumors correlates with poorer survival in multiple cancer types. R
The Th17/Treg Balance: The Most Clinically Relevant Axis
The Th17/Treg relationship is the most important binary in understanding modern chronic inflammatory and autoimmune disease, and it is determined by a single molecular switch: the cytokine environment during naive T cell activation. R
TGF-β alone promotes FoxP3 and iTreg differentiation.
TGF-β plus IL-6 (or IL-21) promotes RORγt and Th17 differentiation.
This means the same cytokine (TGF-β) produces completely opposite immune outcomes depending on whether IL-6 is present. R
IL-6 is pro-inflammatory by nature, produced by macrophages, epithelial cells, adipocytes, and other cells in response to infection, injury, dysbiosis, and obesity.
In chronic inflammatory states where IL-6 is persistently elevated, the Th17/Treg balance tips toward Th17, sustaining inflammation and suppressing regulatory counter-responses. R
The Th17/Treg balance is disrupted in:
- Obesity (adipocyte-derived IL-6 and TNF-α shift balance toward Th17)
- Gut dysbiosis (pathobionts drive IL-6, IL-1β, and IL-23 that expand Th17)
- Metabolic syndrome and type 2 diabetes
- Autoimmune diseases (RA, MS, SLE, IBD, psoriasis, ankylosing spondylitis)
- Recurrent pregnancy loss (elevated Th17/CD4+ Treg ratio during the ovulatory cycle in women with unexplained recurrent pregnancy loss) R
Plasticity as a disease amplifier:
Under inflammatory conditions, nTregs can lose FoxP3 expression and convert to Th17 cells, producing IL-17 instead of suppressing the inflammatory response. R
This Treg-to-Th17 conversion driven by IL-1β and IL-6 has been documented in the synovium of active rheumatoid arthritis patients, in psoriatic skin, and in intestinal lamina propria in IBD. R
It is one of the mechanisms by which an initial infection or inflammatory trigger can initiate self-perpetuating autoimmunity that persists long after the original trigger has resolved.
Th9: The Anti-Tumor And Allergic Paradox
Th9 cells were identified in 2008 as a distinct subset of CD4+ T cells defined by their production of interleukin-9 (IL-9). R
Prior to this, IL-9 was considered a Th2 cytokine, but two independent groups demonstrated that IL-4 combined with TGF-β, rather than promoting Th2, actually drove a distinct IL-9-producing phenotype that suppressed both Th2 and Treg development. R
Polarizing cytokines: IL-4 plus TGF-β (classical Th9 conditions); alternatively, IL-4 plus IL-1β can drive an alternative Th9 phenotype via NF-κB signaling without TGF-β. R
STAT pathway: IL-4 activates STAT6, which induces interferon regulatory factor 4 (IRF4); TGF-β induces PU.1 (an ETS-family transcription factor). R
Master transcription factors: No single master transcription factor has been identified; Th9 differentiation is governed by a network including PU.1, IRF4, STAT6, GATA3, BATF, STAT5, and FOXO1. R R
Signature cytokine produced: IL-9. R
Th9 cells also produce IL-10 and IL-21 in smaller amounts. R
What Th9 cells do:
Th9 cells are the classic example of a subset with completely opposite effects depending on context.
Allergic disease (pathological Th9):
IL-9 activates mast cells, drives eosinophil recruitment, and promotes airway hyperreactivity. R
Th9 cells are elevated in allergic asthma, allergic rhinitis, food allergy, and atopic dermatitis.
Adoptive transfer of Th9-polarized cells into mice produces allergic airway disease that is blocked by anti-IL-9 antibodies. R
Th9 cells also drive colitis via IL-9 receptor signaling in intestinal epithelial cells. R
Anti-tumor immunity (protective Th9):
In solid tumors, particularly melanoma, tumor-specific Th9 cells have potent anti-tumor activity. R
The mechanism: Th9-derived IL-9 activates mast cells and stimulates CTL activity within the tumor microenvironment, driving tumor cell killing.
Increased Th9 cell counts during anti-PD-1 checkpoint inhibitor (nivolumab) treatment were associated with improved clinical responses in metastatic melanoma patients, suggesting Th9 cells may amplify immunotherapy efficacy. R
The alternative IL-4 plus IL-1β pathway for Th9 differentiation (NF-κB-dependent, TGF-β-independent) generates Th9 cells with stronger tumor cytotoxic activity and potentially better clinical utility for cancer immunotherapy than classical Th9 cells. R
Inhibitors of Th9:
IFN-γ, IL-27, and retinoic acid all inhibit IL-9 production from Th9 cells. R
Th22: The Barrier Defender
Th22 cells are a distinct CD4+ subset defined by production of interleukin-22 (IL-22) in the absence of IL-4, IFN-γ, and IL-17. R
This distinguishes them from Th17 cells, which also produce IL-22: in Th22 cells, IL-22 is the primary cytokine and RORγt-driven IL-17 is absent. R
Polarizing cytokines: IL-6 plus tumor necrosis factor-alpha (TNF-α); IL-12 also promotes Th22 differentiation. R
Master transcription factor: Aryl hydrocarbon receptor (AHR), a ligand-activated transcription factor that responds to environmental signals including gut microbiota-derived tryptophan metabolites. R
Note on TGF-β: Unlike most other subsets, TGF-β inhibits Th22 differentiation. R This is one of the key distinctions in the IL-12/IL-23/TGF-β three-dimensional model of human Th differentiation.
What Th22 cells do:
IL-22 is a member of the IL-10 family of cytokines and acts primarily on non-hematopoietic cells at epithelial barrier surfaces (intestine, skin, lung, liver). R
IL-22 promotes epithelial cell proliferation, wound healing, and production of antimicrobial peptides by barrier epithelium. R
Th22 cells accumulate at barrier surfaces (skin and intestinal mucosa) and are particularly important in epidermal immunity and skin tissue remodeling.
Diseases driven by Th22 excess:
- Psoriasis: Th22 cells are a key player alongside Th17 cells; serum IL-22 levels correlate with psoriasis severity. R
- Atopic dermatitis (AD): IL-22 disrupts skin barrier function by suppressing expression of filaggrin and involucrin in keratinocytes.
- Ankylosing spondylitis and axial spondyloarthritis
- MS (IL-22 has been implicated in blood-brain barrier disruption)
The AHR connection:
AHR is activated not only by cytokines but also by environmental ligands: industrial pollutants (dioxins, polycyclic aromatic hydrocarbons (PAHs)), dietary compounds (indoles from cruciferous vegetables), and gut microbiota metabolites (indole derivatives from tryptophan). R
This means Th22 activity is directly responsive to environmental toxin exposure and diet-derived tryptophan metabolite availability, connecting Th22 immune activity to food choices, pollution exposure, and gut microbiota composition.
T Follicular Helper (Tfh) Cells: The Antibody Architects
Tfh cells are the subset specialized to provide help to B cells and orchestrate the germinal center (GC) reaction, which is the process by which B cells undergo somatic hypermutation and affinity maturation to produce high-affinity, class-switched antibodies. R
Without Tfh cells, the immune system cannot generate memory B cells or long-lived plasma cells: all vaccine-induced antibody responses depend on functional Tfh cells. R
Polarizing cytokines: IL-6 and IL-21 initiate Tfh differentiation; ICOS (inducible T cell co-stimulator) signaling is critical for Tfh cell entry into B cell follicles. R
STAT pathway: IL-21 activates STAT3; IL-6 also activates STAT3. R
Master transcription factor: Bcl-6 (B-cell lymphoma 6 protein). R
Bcl-6 drives Tfh differentiation by repressing alternative lineage pathways (Th1, Th2, Th17) and upregulating the chemokine receptor CXCR5, which directs Tfh cells to migrate into B cell follicles. R
Signature molecules: CXCR5, programmed cell death protein 1 (PD-1), ICOS, and IL-21. R
What Tfh cells do:
Upon entry into the GC, Tfh cells engage B cells through CD40-CD40 ligand (CD40L) interactions and IL-21 secretion. R
IL-21 promotes B cell proliferation, class-switch recombination (switching from IgM to IgG, IgA, or IgE), affinity maturation (somatic hypermutation with selection of highest-affinity clones), and plasma cell differentiation. R
Only B cells presenting antigen to Tfh cells with sufficient affinity receive survival signals and continue to mature; lower-affinity B cells are eliminated.
This selection process produces the increasing antibody quality that follows repeated antigen exposure (or vaccination boosters). R
Tfh subsets:
Tfh cells show phenotypic heterogeneity reflecting the cytokine milieu during differentiation:
- Tfh1 cells: Co-express T-bet; produce IFN-γ alongside IL-21; favor IgG2a class switching; relevant in viral infections. R
- Tfh2 cells: Co-express GATA-3; produce IL-4 and IL-13 alongside IL-21; drive IgG1 and IgE class switching; elevated in allergy and atopic disease. R
- Tfh17 cells: Co-express RORγt; produce IL-17 alongside IL-21; prominent in mucosal immune responses. R
- T follicular regulatory (Tfr) cells: FoxP3-expressing Tregs that infiltrate GCs and regulate the magnitude of the GC response by suppressing excessive B cell proliferation. R
Diseases driven by Tfh excess or dysregulation:
- SLE (aberrant GC formation driven by self-reactive Tfh and autoreactive B cells producing anti-dsDNA antibodies)
- RA (synovial Tfh cells promote local autoantibody production)
- Primary Sjogren's syndrome
- Type 1 diabetes (Tfh involvement in anti-islet autoantibody production)
- Allergy: Tfh2 cells drive IgE class switching in B cells at GCs; elevated Tfh activation and GC formation are found in allergic individuals. R
- HIV infection: Tfh cells serve as major reservoirs for HIV in lymphoid tissue. R
Tfh insufficiency:
Reduced Tfh activity impairs vaccine responses and antibody-mediated protection against infections.
Primary immunodeficiencies affecting Tfh development present with dysfunctional germinal centers, poor vaccine antibody responses, and recurrent bacterial infections. R
Cross-Regulation: How The Subsets Talk To Each Other
T helper cell subsets do not operate independently.
Each subset produces cytokines that simultaneously promote its own maintenance and suppress competing lineages. R
Key cross-regulatory interactions:
- IL-12 (Th1 inducer) suppresses Th2 differentiation by inducing T-bet and inhibiting GATA-3 in CD4+ T cells. R
- IFN-γ (Th1 signature cytokine) inhibits Th17 differentiation by suppressing STAT3 activity and RORγt expression. R
- IL-4 (Th2 signature cytokine) inhibits Th1 differentiation by inducing GATA-3 that antagonizes T-bet.
- IL-4 inhibits Th17 differentiation via unknown mechanisms, but the effect is consistent. R
- TGF-β suppresses both Th1 and Th2 but, combined with IL-6, drives Th17; combined with IL-4, drives Th9; and alone promotes Treg. R
- IL-2/STAT5 signaling suppresses both Tfh and Th17 differentiation while promoting Treg maintenance. R
- IL-6 converts nTregs toward Th17 in inflammatory environments, destabilizing FoxP3 expression and promoting RORγt expression in formerly suppressive cells. R
- Retinoic acid (RA) promotes Treg differentiation while inhibiting Th17 differentiation, and inhibits IL-9 production by Th9 cells. R
- FoxP3 directly antagonizes RORγt in cells co-expressing both, such that dominant FoxP3 pushes toward Treg and dominant RORγt pushes toward Th17. IL-6 overcomes FoxP3-mediated RORγt suppression. R
The gut microbiota as a Th balance regulator:
Commensal bacteria directly regulate Th cell balance.
Segmented filamentous bacteria (SFB) are potent inducers of intestinal Th17 responses.
Multiple Clostridia species (Clusters IV and XIVa) produce short-chain fatty acids (SCFAs), particularly butyrate, that promote intestinal iTreg differentiation via HDAC inhibition and FoxP3 CNS1 (conserved non-coding sequence 1) activation. R
Dysbiosis (loss of Clostridia and SCFA-producing bacteria) shifts the mucosal Th17/Treg balance toward Th17, contributing to intestinal inflammation, leaky gut, and systemic immune activation. R
Supplements That Influence Each Subset
The goal of supplementation is not to globally suppress or activate immunity, but to shift the balance: reduce Th17 and Th1 excess in autoimmune and inflammatory contexts, reduce Th2 excess in allergic contexts, and strengthen Treg activity as the common regulatory denominator.
Most supplements discussed here act on multiple subsets simultaneously, which is why mechanism matters more than a simple "take X for Th17" framing.
An important caveat before the list:
A supplement that suppresses Th1 when Th1 is pathologically dominant (autoimmunity, transplant rejection) may blunt an appropriate Th1 response (antimicrobial defense, anti-tumor surveillance) in a different context.
These compounds are immunomodulatory, not unidirectionally immunosuppressive.
Matching the intervention to the immune context matters.
Vitamin D3
Primary effect: Promotes Treg and Th2 differentiation; suppresses Th1 and Th17 differentiation.
Calcitriol (1,25-dihydroxyvitamin D3), the active form of vitamin D, directly inhibits IFN-γ production and reduces T-bet expression in Th1 cells, while simultaneously increasing GATA-3 expression and Th2 cytokine production. R
Calcitriol inhibits Th17 differentiation by reducing RORγt expression and IL-17A production; it promotes Treg differentiation by increasing FoxP3 expression through vitamin D response elements (VDREs) in the FoxP3 gene locus. R
A randomized double-blind placebo-controlled trial in Hashimoto's thyroiditis patients (50,000 IU cholecalciferol weekly for 3 months) produced a significant decrease in the Th17/Tr1 ratio, suggesting a meaningful shift toward regulatory immune balance in an autoimmune context. R
Note: because calcitriol promotes Th2 as well as Treg, vitamin D supplementation in people with Th2-dominant conditions (atopic disease, SLE) requires careful context and may not always be straightforwardly beneficial.
The macrophage-local conversion of 25-OHD3 to calcitriol at sites of inflammation is the most immunologically relevant mechanism in vivo, underscoring the importance of adequate 25-OHD serum levels (above 40 ng/mL). R
Subsets modulated: Th17 (down), Th1 (down), Treg (up), Th2 (up, context-dependent).
Product: Vitamin D3 + K2
Omega-3 Fatty Acids (EPA and DHA)
Primary effect: Suppresses Th1 and Th17; promotes Treg and shifts Th1/Th2 balance toward Th2.
Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) reduce ex vivo Th17 polarization of CD4+ T cells by suppressing the cytokine environment that drives RORγt expression. R
In type 1 diabetes patient-derived peripheral blood mononuclear cells (PBMCs), EPA and DHA rebalanced the Th1/Th2 ratio from approximately 12:1 down to 1:1, while simultaneously reducing Th17 cells and increasing Tregs; omega-6 arachidonic acid (AA) did the opposite, significantly reducing Tregs and elevating Th17 cells. R
Omega-3 fatty acid supplementation reduces IL-17 plasma levels in children with asthma, consistent with Th17 suppression in a Th2/Th17-mixed allergic airway condition. R
EPA and DHA promote Treg accumulation and proliferation in liver, spleen, and adipose tissue in animal models; both fatty acids promote in vitro proliferation of CD4+ CD25+ Treg cells. R
The mechanism involves incorporation of EPA and DHA into immune cell plasma membranes, altering lipid raft organization at the immunological synapse, suppressing TCR signal transduction, and generating specialized pro-resolving lipid mediators (resolvins, protectins, maresins) that actively terminate inflammatory signaling. R
Subsets modulated: Th17 (down), Th1 (down), Treg (up), Th2 (modestly up).
Product: High-dose EPA/DHA fish oil
Curcumin
Primary effect: Suppresses Th17; promotes Treg; modulates Th1/Th2 balance context-dependently.
Curcumin inhibits Th17 differentiation by downregulating the IL-6/STAT3/RORγt signaling axis: in a multiple sclerosis (MS) experimental autoimmune encephalomyelitis (EAE) model, curcumin treatment dramatically decreased IL-17, TGF-β, IL-6, IL-21, STAT3, and RORγt expression, and inhibited STAT3 phosphorylation, reducing Th17-driven disease severity. R
A clinical study on systemic lupus erythematosus (SLE) patients showed that low-dose curcumin (0.1 and 1 μg/ml in culture) decreased Th17 cell percentages and IL-17A production while increasing Treg percentages and TGF-β1 production in CD4+ T cells from SLE patients; these effects were not observed in healthy controls, suggesting curcumin preferentially corrects disease-state imbalance rather than suppressing normal immune function. R
A clinical trial using nano-curcumin in episodic migraine patients significantly reduced serum IL-17 messenger RNA (mRNA), and nano-curcumin in COVID-19 patients decreased Th17 cell numbers and associated inflammatory cytokines in both mild and severe disease. R
Curcumin induces FoxP3 expression in Tregs in graft-versus-host disease (GvHD) models and promotes anti-inflammatory cytokine production from Tregs in IBD and cancer contexts. R
Bioavailability caveat: Standard curcumin powder has very poor oral bioavailability. Phytosome-bound, nanoparticle-encapsulated, or piperine-enhanced formulations are significantly better absorbed.
Subsets modulated: Th17 (down), Treg (up), NF-κB-driven inflammation (down across multiple subsets).
Product: Curcumin phytosome or nano-curcumin
Resveratrol
Primary effect: Reverses Th17/Treg imbalance; modulates Th1/Th2 balance toward Th2 at the intestinal mucosal level.
Resveratrol reverses Th17/Treg imbalance by suppressing the aryl hydrocarbon receptor (AHR) pathway, which normally promotes RORγt expression and Th17 differentiation. R
A clinical trial in immune thrombocytopenic purpura (ITP) patients, a condition characterized by Th17/Treg imbalance, showed that resveratrol treatment reversed the imbalance through AHR pathway suppression, simultaneously reducing IL-17A and IL-22 production and increasing IL-10 and FoxP3 expression. R
In the context of intestinal inflammation and ulcerative colitis (UC), resveratrol shifted both the Th1/Th2 balance toward Th2 (reducing intestinal pro-inflammatory cytokines) and the Treg/Th17 balance toward Treg, improving disease activity indices in animal models. R
Resveratrol also ameliorates dysregulation of Th1, Th2, Th17, and Treg-related transcription factor signaling in neurodevelopmental disorder models, suggesting broad multi-subset immunomodulatory activity. R
Subsets modulated: Th17 (down via AHR suppression), Treg (up via FoxP3 induction), Th1/Th2 balance (context-dependent).
Product: Trans-resveratrol supplement
Butyrate And Short-Chain Fatty Acids (SCFAs)
Primary effect: Strongest known promoter of colonic Treg differentiation; suppresses Th17 at physiological concentrations; modulates Th1 in a concentration-dependent manner.
Butyrate is the most potent SCFA for immune modulation and acts primarily via two mechanisms: (1) inhibition of histone deacetylase (HDAC) enzymes, increasing histone acetylation at the FoxP3 gene locus and promoting Treg differentiation; and (2) signaling through G protein-coupled receptor 109A (GPR109A) on dendritic cells and macrophages, indirectly promoting FoxP3+ Treg expansion. R
SCFAs administered in drinking water to germ-free mice (which have almost no colonic Tregs due to absent microbiota) dramatically increased colonic Treg frequency and FoxP3 + IL-10 expression; this effect was dependent on the GPR43 (FFAR2) receptor and HDAC inhibition. R
Butyrate at low concentrations (0.5 mM range) suppresses Th17 differentiation by downregulating RORγt expression in naive CD4+ T cells undergoing Th17 polarization; at higher concentrations (1 mM), butyrate can also enhance Th1 differentiation via T-bet upregulation and IL-10 co-production, producing an effector Th1 phenotype that can limit colitis. R
In rheumatoid arthritis (RA), butyrate peaks in the early morning (inversely correlating with morning stiffness severity), and clinical acetate levels positively correlate with the IL-17+ CD4+ T cell ratio, suggesting that the SCFA profile directly governs daily fluctuations in Th17 activity. R
How to raise butyrate:
Dietary fiber fermentation by colonic bacteria is the primary source.
Supplemental sodium butyrate or tributyrin (a more stable pro-drug form) bypass the microbiome requirement.
High-fiber diets emphasizing resistant starch, inulin, and pectin feed butyrate-producing Clostridia (Faecalibacterium prausnitzii, Roseburia intestinalis, Eubacterium hallii).
Subsets modulated: Treg (strongly up, colonic), Th17 (down at low doses), Th1 (context and dose-dependent, can increase or decrease).
Sulforaphane
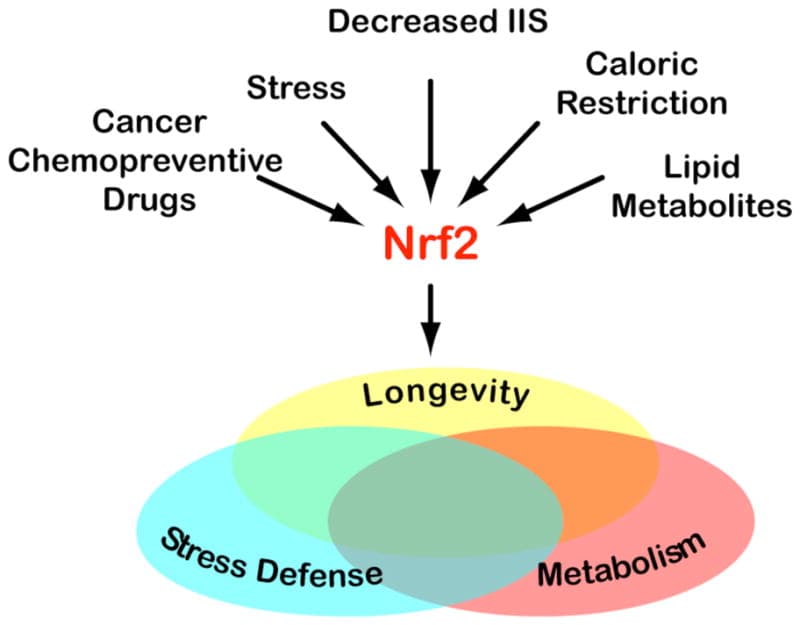
Primary effect: Activates nuclear factor erythroid 2-related factor 2 (Nrf2), which reduces oxidative stress in dendritic cells and supports Th1 immune competence; also suppresses Th17 via indirect NF-κB inhibition.
Sulforaphane (SFN), the isothiocyanate from cruciferous vegetables, activates Nrf2 in dendritic cells, restoring the age-related decline in Th1 immunity. R
Nrf2 activation in antigen-presenting cells (APCs) raises intracellular glutathione (GSH), and elevated GSH in APCs favors Th1 polarization of naive CD4+ T cells; GSH depletion in APCs shifts the response from Th1 to Th2. R
Sulforaphane also inhibits NF-κB signaling and reduces pro-inflammatory cytokine production (IL-6, IL-1β, TNF-α) from macrophages and dendritic cells, indirectly reducing the cytokine milieu that drives Th17 expansion. R
This makes sulforaphane distinct from most other compounds listed here: rather than directly suppressing all T helper cell activity, it specifically supports Th1 competence (which is often lost in aging, immune senescence, and Th2-dominant conditions), while reducing the inflammatory cytokine environment that drives Th17 pathology.
Subsets modulated: Th1 (supports, particularly in aging and immunosenescence context), Th17 (indirectly down via NF-κB/IL-6 reduction), Th2 (partially countered by Th1 restoration).
Product: Sulforaphane from broccoli sprout extract
Berberine
Primary effect: Improves Treg/Th17 balance primarily via gut microbiota modulation; independently inhibits Th17 differentiation via STAT3 suppression.
Berberine (BBR) treatment in a DSS-induced ulcerative colitis mouse model improved Treg/Th17 balance; critically, this effect was abolished when gut microbiota were depleted by antibiotics, and fecal transplantation from berberine-treated mice transferred the Treg/Th17 balance benefit to untreated colitic mice, demonstrating that berberine's immunomodulatory effects are substantially mediated through microbiota remodeling. R
Berberine increases Eubacterium species (butyrate-producers) and Bacteroides while reducing pathobiont Desulfovibrio, shifts that favor colonic Treg induction via the butyrate-HDAC-FoxP3 axis described above. R
Berberine directly inhibits STAT3 phosphorylation, reducing Th17 differentiation independent of the microbiome, through a mechanism similar to its anti-diabetic action via AMPK (AMP-activated protein kinase) activation that suppresses mTORC1-driven T cell metabolic programming required for Th17 differentiation. R
Subsets modulated: Th17 (down, dual mechanism: direct STAT3 + indirect butyrate-mediated), Treg (up via microbiome-butyrate route).
Product: Berberine supplement
Quercetin
Primary effect: Reduces Th2 cytokines in allergic contexts; modulates Th1/Th2 balance and Treg/Th17 balance in inflammatory conditions.
Quercetin reduces IL-4, IL-5, and IL-13 production (the Th2 signature cytokines) in vitro, relevant to its anti-allergic and anti-asthma properties. R
Quercetin improves the imbalance of Th1/Th2 and Treg/Th17 in allergic rhinitis, reducing both Th2-driven IgE responses and Th17-driven neutrophilic inflammation while supporting regulatory immune activity. R
Quercetin inhibits NF-κB and MAPK signaling pathways that drive both Th2 and Th17 effector cytokine production, and activates Nrf2 via a mechanism similar to sulforaphane, supporting antioxidant defenses that favor Th1 competence. R
Subsets modulated: Th2 (down in allergic context), Th17 (down), Th1/Treg (context-dependent support).
Product: Quercetin with bromelain
Probiotics (Lactobacillus, Bifidobacterium, Multi-Strain)
Primary effect: Context-dependent; the most important generalizable effect is Treg expansion via regulatory dendritic cell (rDC) induction and IL-10 / TGF-β promotion.
A mixture of five probiotic strains (Lactobacillus acidophilus, Lactobacillus casei, Lactobacillus reuteri, Bifidobacterium bifidum, and Streptococcus thermophilus) significantly increased CD4+ FoxP3+ Treg populations in mesenteric lymph nodes, decreased Th1, Th2, and Th17 cytokines in CD4+ T cells and B cells, and produced therapeutic effects in experimental IBD, atopic dermatitis, and rheumatoid arthritis models; the mechanism involved generation of regulatory dendritic cells expressing high IL-10, TGF-β, COX-2, and indoleamine 2,3-dioxygenase (IDO). R
Different strains have different dominant effects:
- Immunostimulatory probiotics (Lactobacillus acidophilus, some Lactobacillus casei strains): Promote IL-12 production, stimulate NK cells, and drive Th1 responses, useful for countering Th2-dominant allergy and infections. R
- Immunoregulatory probiotics (Lactobacillus rhamnosus GG, Bifidobacterium longum, multi-strain formulations): Drive Treg expansion and IL-10 production, suppress excessive Th1/Th2/Th17, more relevant to autoimmune and inflammatory contexts. R
- Bifidobacterium bifidum strains specifically support Treg/Th17 plasticity and increase FoxP3+ Treg numbers while inhibiting JAK and NF-κB pathways that drive pro-inflammatory T cell activation. R
Probiotic effects are also substantially mediated through SCFA production, with butyrate-producing strains contributing the most to colonic Treg maintenance (see butyrate section above).
Subsets modulated: Treg (up, most consistent effect), Th1 (strain-dependent: up or down), Th2 and Th17 (generally down with immunoregulatory formulations).
Product: Multi-strain probiotic with Lactobacillus and Bifidobacterium
Summary Table
| Supplement | Th1 | Th2 | Th17 | Treg | Best Use Context |
|---|---|---|---|---|---|
| Vitamin D3 | Down | Up | Down | Up | Autoimmune, Th17-dominant, general |
| Omega-3 EPA/DHA | Down | Modest up | Down | Up | Autoimmune, metabolic inflammation |
| Curcumin (bioavailable) | Neutral | Neutral | Down | Up | Th17-dominant autoimmune, IBD |
| Resveratrol | Neutral | Modest up | Down | Up | Th17/Treg imbalance, IBD |
| Butyrate/SCFAs | Dose-dependent | Neutral | Down (low dose) | Strongly up | Gut dysbiosis, colonic Treg deficit |
| Sulforaphane | Up (supports) | Down (indirect) | Down | Neutral | Aging, immunosenescence, Th2-dominant |
| Berberine | Neutral | Neutral | Down | Up | IBD, metabolic disease, microbiome-mediated |
| Quercetin | Neutral-up | Down | Down | Neutral-up | Allergic/Th2-dominant, Th17 coexistence |
| Probiotics (regulatory strains) | Variable | Down | Down | Up | All inflammatory/autoimmune contexts |
Testing: How To Assess Your T Helper Balance
Direct measurement of T helper cell subset percentages and their associated cytokines is possible, but most tests used clinically are indirect markers rather than true subset counts.
Here is what is available, what each test actually measures, and how to interpret it.
Direct T Cell Subset Testing
Lymphocyte subset panels by flow cytometry are the gold-standard method for directly quantifying T helper cell populations.
These panels measure the percentage and absolute count of CD4+ T cells expressing the surface markers or intracellular cytokines that define each subset.
The markers used:
- Th1: CD4+ IFN-γ+ (intracellular) or CD4+ CXCR3+
- Th2: CD4+ IL-4+ (intracellular) or CD4+ CRTH2+ (CRTh2, the prostaglandin D2 receptor, is a reliable surface Th2 marker)
- Th17: CD4+ IL-17A+ (intracellular) or CD4+ CCR6+ (surface, less specific)
- Treg: CD4+ CD25+ FoxP3+ (intracellular FoxP3 staining required for specificity)
- Tfh: CD4+ CXCR5+ PD-1+ (circulating Tfh, cTfh)
These tests are run in academic medical centers and specialty immunology labs and are typically ordered by immunologists, rheumatologists, or allergy specialists investigating autoimmune or immune deficiency disorders.
Immunosciences Lab and a small number of functional medicine labs offer clinical T helper subset panels directly to patients.
LabCorp and Quest do not routinely offer Th1/Th2/Th17/Treg subset breakdown as a standard panel; they offer basic CD4/CD8 counts (relevant for HIV monitoring) but not subset cytokine profiling.
Cytokine Panels: The Most Practical Proxy
Because direct T cell subset flow cytometry is expensive and not widely available, serum cytokine panels are the most practical proxy for T helper balance in a functional medicine context.
Measuring the cytokines each subset produces gives an indirect picture of which subsets are dominant:
- Th1 activity proxy: IFN-γ, TNF-α, IL-2, IL-12
- Th2 activity proxy: IL-4, IL-5, IL-13, total IgE
- Th17 activity proxy: IL-17A, IL-17F, IL-22, IL-6 (upstream driver)
- Treg activity proxy: IL-10, TGF-β (not a specific marker but correlates with Treg function)
- Th9 activity proxy: IL-9 (uncommonly tested)
- Th22 activity proxy: IL-22 (shares with Th17, context-dependent)
Labs that offer cytokine panels:
- Vibrant Wellness offers a cytokine panel including IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12, IL-13, IL-17A, IFN-γ, TNF-α, and others (accessible through the Cellular Zoomer or standalone cytokine ordering).
- Cyrex Labs Array 11 (cytokine test): IFN-γ, IL-4, IL-1β, TGF-β, and others, developed specifically for autoimmune and neurological immune profiling.
- Great Plains Laboratory offers an organic acids-adjacent immune cytokine panel.
- Quest Diagnostics offers a TGF-β1 test and IL-6 test; broader cytokine panels are available as send-out through specialty labs.
Downstream Biomarkers As Subset Proxies
When direct cytokine or subset testing is not available or affordable, several standard labs serve as reasonable proxies:
Total IgE and allergen-specific IgE:
Elevated total IgE above 100 IU/mL, combined with positive allergen-specific IgE (RAST or ImmunoCAP testing), strongly indicates Th2/Tfh2 overactivity driving IgE class-switching. R
This is the single most widely available indirect Th2 marker.
Eosinophil count (from CBC with differential):
Peripheral eosinophilia above 500 cells/μL correlates with Th2 and Th9 activity; above 1,500 cells/μL suggests more pronounced Th2/ILC2 (innate lymphoid cell type 2) activation or eosinophilic disorder.
Eosinophils are directly driven by IL-5 from Th2 cells.
High-sensitivity C-reactive protein (hsCRP) and erythrocyte sedimentation rate (ESR):
These are non-specific inflammatory markers but elevated hsCRP and ESR in the context of a known autoimmune condition suggest active Th1 and/or Th17-mediated inflammation.
hsCRP above 3 mg/L in the absence of acute infection warrants investigation of Th1/Th17 drivers.
IL-6 serum level:
IL-6 is the pivotal upstream cytokine that shifts TGF-β-mediated Treg differentiation toward Th17 differentiation.
Elevated serum IL-6 (above laboratory reference range, typically >7 pg/mL) in a chronic inflammatory context is a direct signal that the Th17/Treg balance is being actively shifted toward Th17. R
Vibrant Wellness and several specialty labs offer standalone serum IL-6.
Antinuclear antibodies (ANA) and anti-dsDNA:
ANA positivity at 1:160 or higher, particularly with anti-dsDNA antibodies, reflects aberrant Tfh cell activity driving autoreactive B cell antibody production. R
Positive ANA with clinical autoimmune symptoms warrants full autoimmune panel and specialist referral.
Autoantibody panels (anti-CCP, RF, anti-thyroglobulin, anti-TPO, anti-GAD65, etc.):
Each autoantibody set reflects a specific Th1 and/or Tfh-driven autoreactive B cell response in a particular tissue.
Anti-thyroid peroxidase (TPO) and anti-thyroglobulin antibodies: Th1 and Tfh-mediated thyroid autoimmunity (Hashimoto's).
Anti-cyclic citrullinated peptide (anti-CCP) and rheumatoid factor (RF): Th17 and Tfh-mediated synovial autoimmunity.
Anti-GAD65 and anti-islet antibodies: Th1-mediated beta cell destruction in type 1 diabetes.
25-hydroxyvitamin D (25-OHD):
Because vitamin D is one of the most potent physiological modulators of Th17 and Treg balance, serum 25-OHD is an indirect but highly actionable indicator of Th balance potential.
25-OHD below 30 ng/mL is associated with higher Th17 activity and lower Treg counts in autoimmune cohorts.
Target 40 to 60 ng/mL for optimal immunomodulatory effect based on available evidence. R
Recommended Lab Panel For T Helper Assessment
For a functional medicine patient trying to understand their T helper balance, a practical starting panel includes:
- Complete blood count (CBC) with differential (eosinophil count as Th2 proxy)
- hsCRP and ESR (non-specific inflammatory activity)
- IL-6 serum (Th17 upstream driver)
- 25-OHD (vitamin D status, Th17/Treg modulator)
- Total IgE and relevant allergen-specific IgE (Th2/Tfh2 marker)
- ANA screen with reflex (Tfh-driven autoreactive B cell activity)
- IL-17A and IFN-γ serum (direct Th17 and Th1 cytokines; available through Vibrant Wellness or specialty labs)
- IL-10 serum (Treg activity proxy)
This panel does not replace flow cytometric T cell subset measurement, but it gives enough signal to determine the dominant immune phenotype in most clinical scenarios.
The Cellular Zoomer and Immune Zoomer panels at Vibrant Wellness cover several of these markers and add immune cell functional testing and gut barrier integrity markers that contextualize why the T helper balance is where it is.
Mechanisms Of Action
Simple:
- Every Th subset begins as the same naive CD4+ T cell; the cytokine environment during activation determines which master transcription factor gets expressed, which determines which cytokines the cell will produce and which cells it will instruct.
- Th1 cells (T-bet, IFN-γ) fight intracellular pathogens by activating macrophages and cytotoxic T cells.
- Th2 cells (GATA-3, IL-4/IL-5/IL-13) fight parasites via IgE, mast cells, and eosinophils; in the modern world they mostly drive allergic disease.
- Th17 cells (RORγt, IL-17A/F and IL-22) fight extracellular bacteria and fungi by recruiting neutrophils to barrier surfaces; in excess they drive autoimmunity.
- Tregs (FoxP3, IL-10/TGF-β) suppress all other effector populations via cytokines, CTLA-4, IL-2 consumption, adenosine, and cytolysis; nTregs are thymic, epigenetically stable, and self-reactive; iTregs are peripherally induced and less stable under inflammation.
- The Th17/Treg balance is the most clinically relevant immune axis in chronic disease and is flipped toward Th17 by IL-6, which shifts TGF-β-mediated Treg differentiation toward Th17 differentiation.
- Th9 cells (PU.1/IRF4, IL-9) are paradoxical: they drive allergy and colitis via mast cell activation, but also drive anti-tumor immunity in melanoma and potentiate checkpoint inhibitor responses.
- Th22 cells (AHR, IL-22) defend and repair barrier surfaces; their activity is directly influenced by environmental toxin exposures and gut microbiota tryptophan metabolism.
- Tfh cells (Bcl-6, CXCR5, IL-21) orchestrate all germinal center reactions; without them, no vaccine works and no high-affinity antibody forms. In excess or when autoreactive, they drive SLE, Sjogren's, and other autoantibody-mediated diseases.
Advanced:
- The STAT-to-master-transcription-factor cascade: Each polarizing cytokine binds its receptor and activates a JAK (Janus kinase) that phosphorylates a specific STAT protein. STAT4 (activated by IL-12) drives T-bet expression for Th1. STAT6 (activated by IL-4) drives GATA-3 for Th2. STAT3 (activated by IL-6, IL-21, IL-23) drives RORγt for Th17 and Bcl-6 for Tfh. STAT5 (activated by IL-2) promotes FoxP3 for Tregs and suppresses IL-17 gene expression by directly binding the IL-17 promoter. R The master transcription factors then drive expression of subset-specific cytokine genes while epigenetically silencing competing lineage programs through chromatin remodeling.
- The Th17/Treg molecular switch in detail: TGF-β activates SMAD2/3 transcription factors. SMAD3 binds the FoxP3 CNS1 enhancer element (a TGF-β-NFAT response element), driving FoxP3 transcription and iTreg differentiation. IL-6, acting through STAT3, simultaneously induces RORγt and suppresses SMAD3-mediated FoxP3 activity. R IL-6-activated STAT3 also upregulates SOCS3 (suppressor of cytokine signaling 3), which provides negative feedback on STAT3 and modulates the kinetics of Th17 expansion. R At the chromatin level, Th17 differentiation requires an open chromatin conformation at the IL-17 locus driven by IRF4-BATF complexes, while TGF-β keeps the IL-17 locus closed in Treg-inducing conditions. R
- FoxP3 locus epigenetics and Treg stability: The Foxp3 gene contains three conserved non-coding sequences (CNS1, CNS2, CNS3). CNS3 is essential for nTreg development in the thymus. CNS1 contains the TGF-β-NFAT response element and is required for iTreg induction in the periphery; CNS1-deficient mice develop spontaneous colitis. CNS2 regulates heritable FoxP3 expression through a CpG island (the TSDR) that is fully demethylated in thymic nTregs but partially methylated in iTregs. R The differential methylation of CNS2-TSDR is the molecular basis for the difference in stability between nTregs (stable FoxP3 expression) and iTregs (FoxP3 expression subject to loss under inflammatory conditions), and it is why iTregs can convert to Th17 cells when exposed to IL-6 and IL-1β in vivo.
- AHR as the environmental link in Th22 and Th17: The aryl hydrocarbon receptor (AHR) is a ligand-dependent transcription factor that integrates environmental signals into immune cell fate decisions. AHR activation by dioxins and other industrial pollutants can drive IL-17 and IL-22 expression, generating both Th17 and Th22 activity. R Dietary indoles from cruciferous vegetables produce indole-3-carbinol and its derivative DIM (diindolylmethane), which are AHR ligands that preferentially promote IL-22 production (Th22 activity) over IL-17. Gut microbiota produce indole-3-aldehyde and indole-3-propionic acid from tryptophan, which are also AHR agonists that support mucosal barrier IL-22 activity. This is the mechanistic connection between cruciferous vegetable intake, a diverse tryptophan-metabolizing microbiome, and mucosal barrier integrity.
Genetics
Most T helper subset dysregulation in human disease is polygenic: many variants with small individual effects combine with environmental and microbiome factors to shift the Th balance. Monogenic defects are rare but mechanistically illuminating.
FoxP3 (X-linked):
Loss-of-function mutations cause IPEX syndrome (immune dysregulation, polyendocrinopathy, enteropathy, X-linked), an X-linked condition characterized by neonatal or early infantile onset of autoimmune enteropathy, type 1 diabetes, eczema, thyroiditis, and hemolytic anemia, uniformly fatal without hematopoietic stem cell transplantation. R Demonstrates the absolute non-redundancy of nTreg-mediated self-tolerance.
STAT3 (gain-of-function mutations):
Somatic or germline gain-of-function mutations in STAT3 drive excessive Th17 and Tfh activity, producing a syndrome of multi-organ autoimmunity, cytopenias, lymphoproliferation, and growth failure. The gain-of-function phenotype is the opposite of STAT3 loss-of-function (hyper-IgE syndrome), which impairs Th17 differentiation and produces recurrent Candida and bacterial infections, elevated IgE, eczema, and skeletal abnormalities. R
IL-17 pathway defects:
Loss-of-function variants in IL17A, IL17F, IL17RA, IL17RC, ACT1 (the IL-17 signal adapter), or STAT3 (in its Th17-driving capacity) produce chronic mucocutaneous candidiasis (CMC), confirming that Th17 activity is non-redundantly required for antifungal mucosal immunity in humans. R
Common variants contributing to Th17/Treg imbalance:
- IL23R variants: The IL-23 receptor gene contains protective variants (particularly Arg381Gln, rs11209026) strongly associated with reduced Crohn's disease, psoriasis, and ankylosing spondylitis risk; the protective allele impairs IL-23-driven Th17 expansion. R
- HLA region: The strongest genetic associations with most autoimmune diseases localize to the HLA locus, which determines which peptide antigens can be presented to T cells; specific HLA alleles favor presentation of autoantigens to Th1 and Th17 cells.
- PTPN22 (rs2476601): A gain-of-function variant in protein tyrosine phosphatase N22 (PTPN22), which modulates TCR and B cell receptor signaling thresholds; associated with RA, T1D, SLE, and Graves' disease; likely shifts the balance toward autoreactive Th1 and Th17 activation.
More Research
- The concept of a single naive CD4+ T cell being permanently committed to one subset upon activation is increasingly being replaced by a model of T helper cell plasticity where subset identity is a probability distribution influenced by cumulative environmental signals rather than a binary state. R Cells in clinical tissues often co-express transcription factors from multiple lineages (for example, Foxp3 and RORγt co-expressing cells in the intestinal lamina propria are common), and the relative dominance of these factors determines the functional output.
- The role of the microbiome in Th balance is one of the most active research areas in immunology. The composition of the gut microbiota directly determines local Th17/Treg ratios in the intestinal mucosa, and dysbiosis is a consistent upstream factor in autoimmune disease across multiple independent cohorts. Restoration of microbiota diversity (through dietary fiber, probiotics, or fecal transplant) shifts the Th17/Treg ratio back toward Treg dominance in mouse models and early human trials. R
- Tfh cells are now understood to be a major HIV reservoir in lymphoid tissue; HIV-infected Tfh cells persist under antiretroviral therapy because PD-1 expression on Tfh cells inhibits their CTL-mediated killing. Targeting the PD-1/PD-L1 axis in HIV treatment is under investigation partly to expose infected Tfh cells to CTL surveillance. R
- Vitamin D and the Th balance: Calcitriol (1,25-dihydroxyvitamin D3, the active vitamin D metabolite) promotes iTreg differentiation and suppresses Th17 differentiation through multiple mechanisms including direct activation of FoxP3 gene transcription via vitamin D response elements (VDREs) in the FoxP3 promoter. Vitamin D deficiency is associated with higher Th17 activity and lower Treg counts in autoimmune cohorts.
- The distinction between nTreg and iTreg stability has direct clinical implications for cell therapy: first-generation Treg cell therapy trials using ex vivo-expanded nTregs in graft-versus-host disease and transplant rejection have shown promise, but converting conventional T cells into iTregs for therapy has been hampered by the methylation state of the FoxP3 TSDR making them vulnerable to reprogramming toward Th17 in the inflammatory environments of the target disease. R
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Quercetin
500mg 2x/day
Vitamin D3 + K2
5000 IU + 200mcg/day
DAO Enzyme
1 cap before meals