Iron Overload and Ferritin Dysregulation: The HFE Gene, Hemochromatosis, and Why High Ferritin Isn't Always Inflammation
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.
Iron overload is one of the few chronic conditions where the body has no efficient way to excrete the excess, so it accumulates for decades before it shows up as organ damage.
In this post, we will discuss how iron is normally regulated, the HFE gene mutations behind hereditary hemochromatosis, why ferritin is a confounded biomarker, the Fenton reaction that makes stored iron dangerous, and the practical workup and treatment.

Basics Of Iron Overload And Ferritin
Iron is essential and toxic in the same molecule, which is the central problem your body has to manage.
You need it for oxygen transport, mitochondrial energy production, and DNA synthesis, but free iron catalyzes the formation of the most destructive radical in human biology.
Iron overload is the state of having too much total body iron, and unlike almost every other nutrient, humans have no regulated pathway to excrete it.
Absorption is the only control point, so once iron is in, it stays in, distributed between a safe storage form and a dangerous reactive pool.
Ferritin is the intracellular protein shell that stores iron in its safe, sequestered form, a 24-subunit cage holding up to roughly 4,500 iron atoms per molecule. R
A small amount of ferritin leaks into the blood, and that serum ferritin is what most labs measure as a proxy for total iron stores.
The problem this post keeps returning to is that serum ferritin rises for two completely different reasons, real iron loading and inflammation, and telling them apart changes everything about what you do next.
How Iron Is Normally Regulated
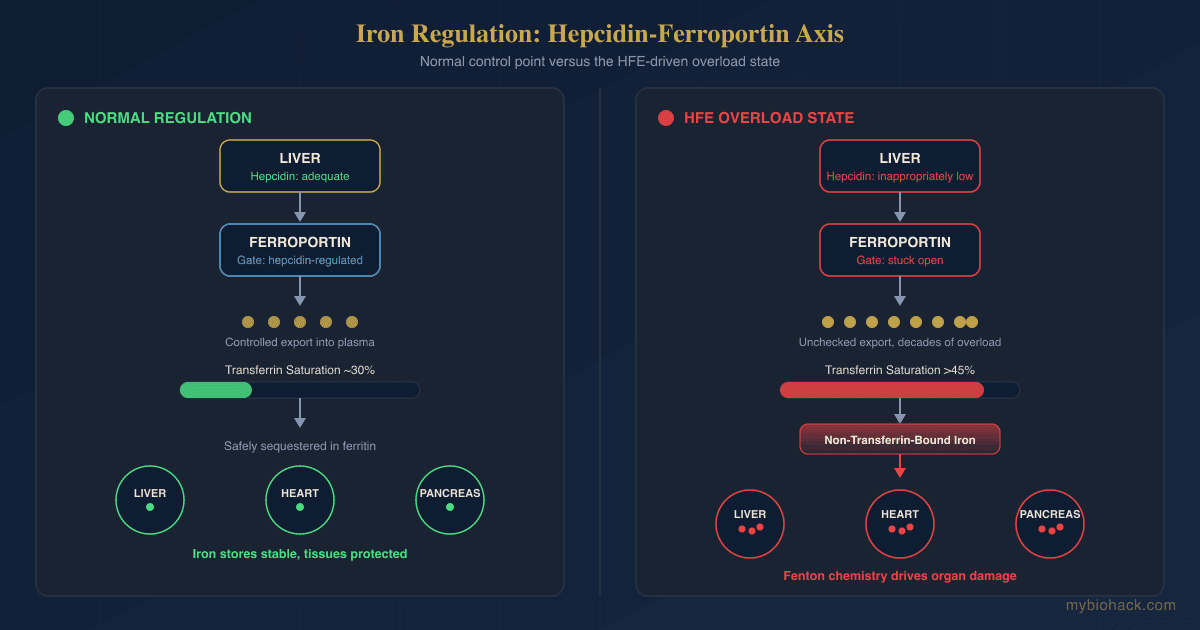
Iron regulation runs on a single master hormone, and understanding it makes hereditary hemochromatosis obvious.
Hepcidin is a 25 amino acid peptide hormone made by the liver, and it is the central regulator of how much iron enters your bloodstream. R
Hepcidin works by controlling a single exporter protein called ferroportin, which is the only known channel that moves iron out of cells and into the blood. R
Ferroportin sits on the surface of the duodenal cells that absorb dietary iron, on macrophages that recycle iron from old red blood cells, and on liver cells that store it. R
When iron stores are high, the liver makes more hepcidin, hepcidin binds ferroportin and triggers its internalization and degradation, and iron export drops. R
When iron is needed, hepcidin falls, ferroportin stays on the cell surface, and more iron flows into circulation.
Two signals can override the iron sensor, and the main one is erythroferrone, a hormone released by developing red cell precursors that suppresses hepcidin whenever the marrow needs iron to build hemoglobin. R
Inflammation pushes the opposite way, raising hepcidin through interleukin-6 and STAT3 signaling, which locks iron inside cells and is the mechanism behind the anemia of chronic disease. R
Transferrin is the blood protein that carries iron once it is exported, and it normally runs about a third saturated, keeping the amount of free, unbound iron close to zero.
The upstream sensors that tell the liver how much iron is present include the HFE protein, transferrin receptor 2 (TfR2), and hemojuvelin (HJV), which together tune hepcidin production to iron status. R
This is the entire system in one sentence, iron sensors raise hepcidin, hepcidin lowers ferroportin, and ferroportin controls how much iron gets in.
The HFE Gene And Hereditary Hemochromatosis
Hereditary hemochromatosis is an inherited disorder of iron overload, and the most common form is caused by mutations in the HFE gene.
The HFE protein is one of the sensors that signals iron status to the hepcidin machinery, so when it is defective, the liver misreads iron stores as low.
The result is inappropriately low hepcidin despite systemic iron overload, which keeps ferroportin active and lets the gut keep absorbing iron long after stores are full. R

Two HFE variants matter most, C282Y (rs1800562) and H63D (rs1799945), and they behave very differently.
C282Y is the high-risk allele, and homozygosity for it (two copies) accounts for the large majority of clinically significant hereditary hemochromatosis. R
The C282Y homozygous genotype is common in people of Northern European descent, occurring in roughly 1 in 200 individuals of Anglo-Celtic ancestry. R
Here is the part most people get wrong, carrying the genotype does not mean you will get the disease.
The clinical penetrance of C282Y homozygosity is much lower than its prevalence, with possibly fewer than 5% developing overt clinical iron-overload disease. R
The compound heterozygous genotype (one C282Y plus one H63D) is a low-penetrance state, and these individuals generally do not develop progressive disease unless a cofactor like alcohol, obesity, diabetes, or fatty liver is also present. R
This is why genetics alone never makes the diagnosis, you confirm iron overload with iron studies, and the genotype only tells you the mechanism.
Why Ferritin Is A Confounded Biomarker
This is the section that changes how you read your own labs.
Serum ferritin is used as a proxy for iron stores, but it is also an acute phase reactant, meaning it rises as part of the inflammatory response independent of how much iron you actually have. R
Ferritin goes up nonspecifically in infection, autoimmune disease, chronic kidney disease, metabolic syndrome, liver injury, and malignancy, none of which is iron overload. R
In the presence of inflammation, serum ferritin no longer tracks iron availability, so a high number can reflect a flare rather than a full tank. R
The reverse trap is just as important, inflammation can push a genuinely iron-deficient person's ferritin up into the normal range and hide the deficiency, because the same IL-6 driven hepcidin rise that sequesters iron also drives ferritin synthesis. R
This is why ferritin should never be interpreted alone.
The single most useful companion test is transferrin saturation, calculated from serum iron and total iron binding capacity, because it reflects the iron actually in transit rather than the storage protein. R
In hereditary hemochromatosis, transferrin saturation is characteristically high, often the first marker to rise, whereas in inflammation-driven high ferritin the saturation is typically normal or low.
A high ferritin with a high transferrin saturation points toward iron loading, while a high ferritin with a normal or low saturation points toward inflammation.
Checking a C-reactive protein at the same time helps, because a clearly elevated CRP flags an acute phase response that is inflating the ferritin. R
The practical rule is that ferritin is a screen, not a verdict, and it always needs saturation plus an inflammatory marker before you act on it.

The Fenton Reaction And Iron-Driven Oxidative Damage
The reason iron overload damages organs comes down to one chemical reaction.
The Fenton reaction is the process by which ferrous iron (Fe2+) reacts with hydrogen peroxide to generate the hydroxyl radical, the most reactive and destructive oxygen radical in biology. R
Hydrogen peroxide on its own is relatively mild, but in the presence of free iron it is converted into hydroxyl radicals that damage DNA, oxidize proteins, and peroxidize membrane lipids indiscriminately. R
When iron stores exceed the capacity of transferrin and ferritin to sequester them, a pool of non-transferrin-bound iron (NTBI) appears in the blood and tissues, and this reactive iron is the substrate for Fenton chemistry. R
The hydroxyl radicals generated this way attack polyunsaturated fatty acids in cell membranes, producing lipid peroxides that propagate in a chain reaction and can trigger a specific iron-dependent form of cell death called ferroptosis. R
This is the mechanistic bridge between a lab value and organ failure, excess iron means more NTBI, more NTBI means more hydroxyl radicals, and sustained radical load means cumulative damage to the liver, heart, pancreas, and joints.
It also explains why the same hydroxyl radical chemistry drives oxidative DNA damage, the kind measured by markers like 8-OHdG.
Your antioxidant defenses, especially the NRF2 pathway and glutathione system, are what stand between stored iron and this damage, which is why redox capacity matters as much as the iron number itself. R
Iron Overload And Overlapping Conditions
Iron deposits preferentially in specific organs, and the clinical picture follows where it lands.
Iron overload target organs and consequences (not an exclusive list):
- Heart (iron-loading cardiomyopathy with heart failure and arrhythmia, the second most common fatal complication of untreated hemochromatosis) R
- Joints (arthropathy, classically the second and third knuckles and the ankles, often an early and persistent complaint) R
- Liver (the most common site of damage, progressing from elevated enzymes to fibrosis to cirrhosis, with 20 to 30% of cirrhotics developing hepatocellular carcinoma) R
- Pancreas (beta-cell damage producing the classic "bronze diabetes" of pigmentation plus glucose intolerance) R
- Pituitary and gonads (hypogonadism, low libido, and fatigue from iron deposition in the endocrine axis) R
The risk of cirrhosis is not evenly distributed, and ferritin predicts it well at the extremes.
Among people with phenotypic hemochromatosis, a serum ferritin above 1,000 ng/mL is strongly associated with advanced hepatic fibrosis, while values under that threshold rarely show cirrhosis. R
Beyond frank hemochromatosis, elevated body iron stores have been linked epidemiologically to insulin resistance, fatty liver, and cardiovascular risk, though the strength of that link is genuinely contested. R
Not all iron overload is genetic, and the most common acquired form is dysmetabolic iron overload syndrome, a moderate iron loading that travels with fatty liver, central obesity, and insulin resistance and presents as high ferritin with normal or only mildly elevated transferrin saturation. R
The other major acquired route is transfusional, since each unit of packed red cells carries 200 to 250 mg of iron and people on chronic transfusion for thalassemia or myelodysplastic syndrome accumulate a burden the body has no way to clear. R
There is a big MAYBE here, some prospective cohorts find a borderline association between iron stores and coronary heart disease, while others, including the ARIC analysis of ferritin and LDL oxidation, find none. R R
The honest read is that overt hemochromatosis is unambiguously harmful, while the risk of high-normal iron in an otherwise healthy person is real in mechanism but modest and inconsistent in the epidemiology.
How To Reduce Iron Overload
The only proven way to remove iron from the body is to remove blood, and everything else is supportive.
1. Therapeutic Phlebotomy
Removing a unit of blood forces the body to pull stored iron out of ferritin to build new red cells, steadily depleting the iron burden.
This is the first-line, standard-of-care treatment for hereditary hemochromatosis, typically 500 mL removed weekly until serum ferritin falls to the low end of normal (often targeted below 50 ng/mL), then spaced out for maintenance. R
Phlebotomy improves fatigue, joint pain, and liver enzymes, and can regress fibrosis in a subset of patients, and treated individuals with hemochromatosis have essentially normal life expectancy if depleted before cirrhosis develops. R
For people with high-normal iron who are not hemochromatotic, regular blood donation accomplishes the same iron reduction and is the simplest intervention available. R
Outside classic hemochromatosis the benefit is less certain, and phlebotomy trials in fatty liver disease conflict, with one randomized iron-depletion trial in hyperferritinemic NAFLD improving liver histology and enzymes while a separate controlled trial found no change in hepatic fat or insulin resistance. R R
2. Reduce Dietary Iron Loading
You cannot chelate your way out of a diet that keeps refilling the tank, so meal composition matters.
Non-heme iron absorption is strongly modifiable, and pairing iron-rich meals with inhibitors rather than enhancers meaningfully lowers uptake. R
Coffee and tea polyphenols, calcium, and phytate from whole grains and legumes all inhibit non-heme iron absorption when taken with meals. R
3. Iron-Binding Polyphenols And Chelators
Several food-derived compounds bind iron in the gut or reduce its reactivity, and while they do not replace phlebotomy, they are reasonable adjuncts.
This is the supplemental form of phytate, a potent non-heme iron chelator in the digestive tract. R
A flavonoid that chelates iron and dampens iron-catalyzed oxidative stress (also a mast cell stabilizer, useful if histamine issues coexist). R
Tea catechins bind non-heme iron and are among the strongest dietary absorption inhibitors (take with iron-containing meals, not away from them). R
An iron chelator that also supports the NRF2 antioxidant response, addressing both the iron and the downstream oxidative arm (can lower ferritin, monitor if you are also iron deficient). R
4. Support The Liver And Antioxidant Defense
Because the damage is oxidative, reinforcing redox capacity protects the organs while iron is being unloaded.
Hepatoprotective and mildly iron-chelating, a sensible adjunct where liver enzymes are elevated. R
A glutathione precursor that shores up the primary defense against hydroxyl-radical damage. R
What To Stay Away From
The fastest way to make iron overload worse is to keep adding iron or to make the iron you have more reactive.
Avoid in iron overload (not an exclusive list):
- Alcohol (synergistic with iron in driving liver fibrosis, and a recognized cofactor that pushes low-penetrance genotypes into overt disease) R
- High-dose vitamin C with meals (ascorbic acid both boosts non-heme iron absorption and mobilizes stored iron into the reactive pool, worsening Fenton chemistry) R
- Iron supplements and iron-fortified foods (unnecessary iron loading, check multivitamins and fortified cereals for added iron)
- Raw shellfish (Vibrio vulnificus thrives on available iron and causes fulminant, sometimes fatal, infection in iron-overloaded people) R
- Uncontrolled heme iron intake (heme iron from red meat is absorbed efficiently and bypasses most dietary inhibitors, so quantity matters)
Testing
Diagnosis rests on two blood markers first, then genetics to explain the mechanism, then imaging if damage is suspected.
Blood And Urine Markers
Serum ferritin: the storage-iron screen, but remember it is an acute phase reactant, so interpret it alongside inflammation. R
I use the Ferritin test (Quest via Fullscript) as the starting screen for iron stores.
Transferrin saturation: the more specific marker for iron loading, calculated from serum iron and TIBC, and typically the first value to rise in hereditary hemochromatosis.
I use the Iron + Total Iron Binding Capacity (TIBC) panel (Quest via Fullscript) to calculate transferrin saturation, and a fasting saturation above 45% warrants genetic follow-up. R
Serum iron alone: useful within the saturation calculation but not interpretable in isolation.
The Iron, Total test (Quest via Fullscript) covers this where only the serum iron value is needed.
Genetic Testing
HFE genotyping: confirms the mechanism once iron studies are abnormal, identifying C282Y and H63D status, but it does not by itself diagnose disease given the low penetrance.
I use the Hereditary Hemochromatosis DNA Analysis (Quest via Fullscript) to determine C282Y and H63D genotype after iron studies are elevated.
Functional Lab Panels
I use the Foundation Zoomer (Vibrant Wellness) to capture the CBC, liver enzymes (AST, ALT, GGT, ALP), and fasting glucose together, which surface the earliest organ signals of iron loading.
For the metabolic overlap (insulin resistance and the "bronze diabetes" picture), I use the Cardio Zoomer (Vibrant Wellness) to assess fasting insulin and metabolic markers alongside lipids.
Because iron and copper metabolism are linked through ceruloplasmin, the copper and zinc status on the Nutrient Zoomer (Vibrant Wellness) can add context, and I cover that interplay in the zinc, copper, and ceruloplasmin post.
Imaging
Liver MRI (R2/T2* iron quantification): the noninvasive gold standard for measuring hepatic iron concentration, largely replacing liver biopsy except where fibrosis staging is needed. R
Cardiac T2* MRI: quantifies myocardial iron directly and predicts heart failure and arrhythmia far better than serum ferritin, with values below 10 ms marking the highest-risk hearts. R
For personalized interpretation of an elevated ferritin or ambiguous iron panel, a consultation can help sort iron loading from inflammation before you commit to phlebotomy.
Mechanisms Of Action
Simple:
- Your body can absorb iron but cannot excrete it, so a broken iron sensor lets the gut keep loading iron for decades until it spills into organs and rusts them from the inside.
Advanced:
- Hepcidin-ferroportin axis failure The HFE protein, together with TfR2 and HJV, normally signals hepatic iron status to upregulate hepcidin transcription through BMP/SMAD signaling. Loss-of-function HFE variants blunt this signal, hepcidin stays inappropriately low, ferroportin remains on the enterocyte and macrophage surface, and iron efflux into plasma continues unchecked despite full stores. R
- Transferrin saturation and NTBI generation As iron efflux outpaces transferrin's binding capacity, saturation climbs and non-transferrin-bound iron appears. NTBI is taken up by hepatocytes, cardiomyocytes, and pancreatic beta cells through ZIP14 and calcium channels, concentrating reactive iron in exactly the organs that fail. R
- Fenton chemistry and lipid peroxidation Intracellular labile Fe2+ reduces hydrogen peroxide to the hydroxyl radical, which abstracts hydrogen from membrane polyunsaturated fatty acids to initiate a self-propagating lipid peroxidation chain. When glutathione peroxidase 4 capacity is exceeded, the accumulation of lipid hydroperoxides triggers ferroptotic cell death, linking iron burden directly to organ-specific tissue loss. R
Genetics
Five genes converge on the same endpoint, insufficient hepcidin for the body's iron load, and HFE is only the most common. R
HFE: Highest Population Risk
HFE encodes an MHC class I-like protein that helps sense transferrin-bound iron and regulate hepcidin.
Loss-of-function variants reduce hepcidin signaling and cause the most common (type 1) hereditary hemochromatosis.
rs1800562: the C282Y variant, disrupts HFE folding and cell-surface presentation, and homozygosity is the primary genetic cause of clinical iron overload. R
rs1799945: the H63D variant, a milder change that rarely causes overload alone but can contribute in compound heterozygotes with a cofactor. R
HJV (HFE2)
HJV encodes hemojuvelin, a BMP co-receptor that strongly upregulates hepcidin.
Mutations cause type 2A juvenile hemochromatosis, a severe early-onset form with rapid iron loading. R
HAMP
HAMP encodes hepcidin itself, the master iron-regulatory hormone.
Loss-of-function mutations abolish hepcidin production and cause type 2B juvenile hemochromatosis, which can be lethal before the fourth decade if untreated. R
TFR2
TFR2 encodes transferrin receptor 2, another sensor of iron-loaded transferrin that helps set hepcidin levels.
Mutations cause type 3 hemochromatosis, generally milder and later-onset, similar to the HFE form. R
SLC40A1
SLC40A1 encodes ferroportin, the iron exporter that hepcidin acts on.
Mutations cause type 4 (ferroportin disease), which comes in two flavors, loss-of-function producing macrophage iron loading, and gain-of-function (hepcidin-resistant) producing a classic hemochromatosis pattern. R
TMPRSS6
TMPRSS6 encodes matriptase-2, which suppresses hepcidin, so it acts in the opposite direction of the hemochromatosis genes.
rs855791: a common variant that influences hepcidin, iron status, and hemoglobin at the population level, and modifies iron handling rather than causing overload outright. R
More Research
Ceruloplasmin ties iron to copper, because this copper-dependent ferroxidase is required to load iron onto transferrin, and functional copper deficiency can produce an iron-handling picture that mimics parts of overload. See the zinc, copper, and ceruloplasmin post for that axis.
Elevated ferritin belongs in the same mental category as an elevated homocysteine, a widely measured marker whose causal weight in cardiovascular disease is real in mechanism but genuinely debated in the outcome data. R
Ferroptosis, the iron-dependent cell death pathway, is an active research front connecting iron biology to neurodegeneration, ischemia-reperfusion injury, and cancer, and it reframes iron overload as a chronic ferroptotic pressure rather than a static storage problem. R
Penetrance is the recurring theme worth internalizing, because carrying a hemochromatosis genotype is common while developing the disease is not, and cofactors like alcohol, obesity, fatty liver, and diabetes are what tip a genotype into a phenotype. R
Phlebotomy evidence is strong for symptom and biochemical improvement but thinner than most assume for hard endpoints, since randomized trials against no treatment are not ethical, so the survival data come from cohorts rather than controlled trials. R
For biomarker tracking over time, I use the Ferritin and Iron + TIBC tests together, because watching the trend in both storage iron and transferrin saturation is far more informative than any single snapshot.
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Methylated B Complex
1 cap/day with food
SAMe
400mg on empty stomach
Resveratrol
250mg/day