Itaconate: The Immunometabolite That Switches Off Inflammation
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.
Itaconate is a small molecule your activated macrophages build from a Krebs cycle intermediate, and it is one of the most powerful brakes the immune system has on its own inflammation.
In this post, we will discuss what itaconate is, how it is made, the four main ways it shuts down inflammatory signaling, its antimicrobial role, why it matters in chronic illness and post-viral disease, the research-tool analogs (4-octyl itaconate and dimethyl itaconate), why there is no clean human supplement yet, how to support the endogenous pathway, testing, mechanisms, and genetics.

What Itaconate Is
Itaconate is an immunometabolite, a metabolite that doubles as a signaling molecule the immune system uses to control itself.
It is not a vitamin, a herb, or a classic supplement.
It is a dicarboxylic acid your own cells synthesize on demand when an immune cell is activated.
For decades itaconate was treated as an industrial chemical used to make plastics and resins, with no known role in human biology.
That changed when researchers found it is one of the most highly induced metabolites in macrophages (the front-line immune cells that engulf pathogens and debris) after they sense an infection. R
When a macrophage detects Lipopolysaccharide (LPS), the endotoxin shed by gram-negative bacteria, itaconate levels inside that cell can rise to become one of its single most abundant metabolites. R
The body does not waste energy producing something at that scale unless it does something important.
What it does is act as a built-in off-switch, a way for an inflamed cell to limit the collateral damage of its own response.
This is the same logic behind Jacob's framing of inflammation as the first stage of the wound healing cycle rather than the root problem, where the real question is what keeps you stuck in inflammation instead of progressing to repair.
How Itaconate Is Made
Itaconate is produced directly off the Krebs cycle (the central energy-producing cycle in your mitochondria).
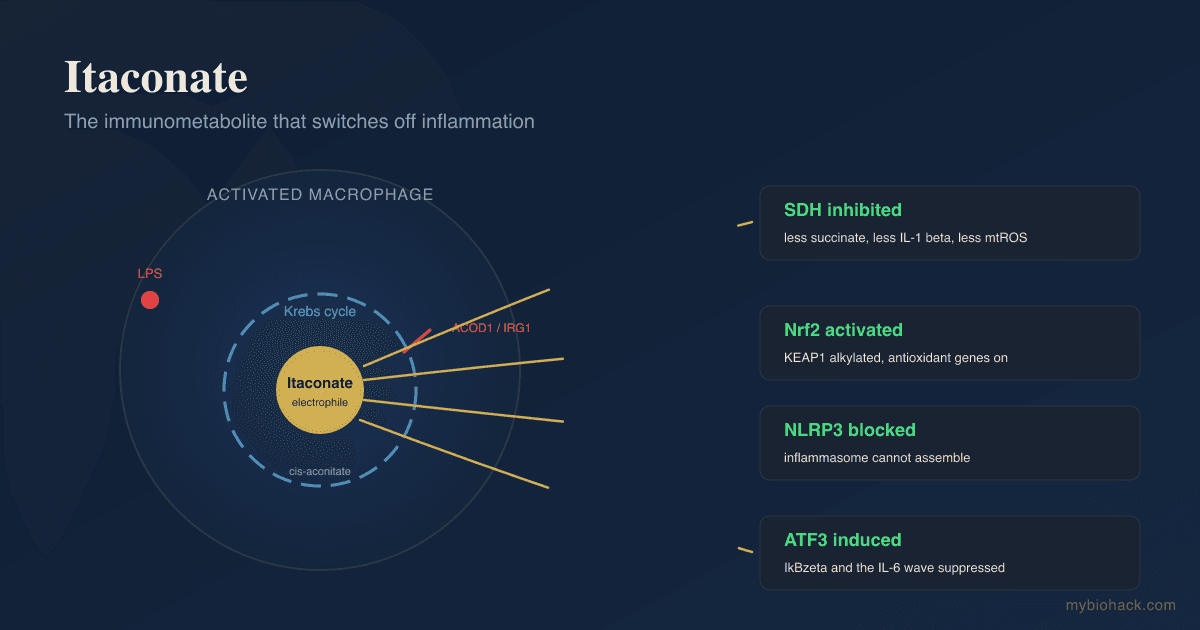
The enzyme that makes it is Aconitate Decarboxylase 1 (ACOD1), encoded by the gene historically called Immunoresponsive Gene 1 (IRG1). R
You will see both names in the literature, and they refer to the same enzyme.
ACOD1 takes cis-aconitate, an intermediate of the Krebs cycle, and removes a carboxyl group from it to form itaconate. R
This only happens to a meaningful degree in activated immune cells of the myeloid lineage, mostly macrophages and monocytes, which is why itaconate is considered an immune-specific metabolite. R
When a macrophage is activated, its Krebs cycle does not just keep spinning to make energy.
It "breaks" at two points so that intermediates can be diverted into signaling roles, and the itaconate detour is one of those breaks.
The diversion of cis-aconitate into itaconate is part of the broader metabolic rewiring that switches an inflammatory macrophage from quiet oxidative metabolism into a glycolytic, pro-inflammatory state, and then helps wind it back down.
How Itaconate Brakes Inflammation
Itaconate is electrophilic, meaning it reacts with and chemically modifies specific cysteine residues on target proteins, a process called alkylation.
This single chemical property explains most of its anti-inflammatory effects, because it lets one small molecule reprogram several different signaling proteins at once.
There are four main brakes, and they overlap.
Succinate dehydrogenase inhibition
The first mechanism discovered was inhibition of Succinate Dehydrogenase (SDH), the enzyme that oxidizes succinate in the Krebs cycle. R
In an inflamed macrophage, succinate accumulates and acts as a danger signal that drives production of the inflammatory cytokine IL-1 beta.
Itaconate inhibits SDH, which lowers the succinate-driven inflammatory signal and reduces reactive oxygen species coming out of the mitochondria. R
Mice that cannot make itaconate (Irg1 knockouts) show higher succinate, higher mitochondrial Reactive Oxygen Species (ROS), and worse inflammation, which is the cleanest evidence that endogenous itaconate is a genuine brake and not just a byproduct. R
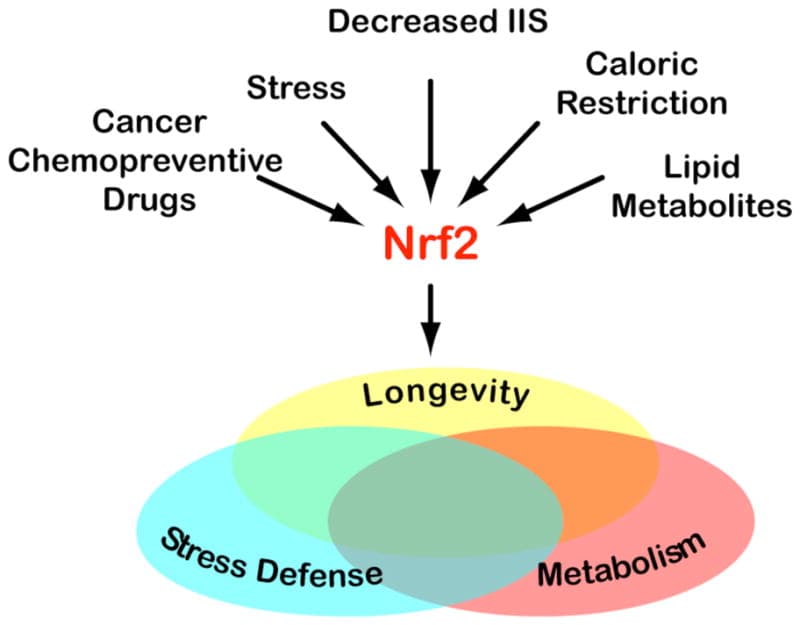
Nrf2 activation via KEAP1 alkylation
The second mechanism is activation of Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2), the master regulator of the antioxidant response, encoded by the gene NFE2L2.
Itaconate directly alkylates cysteine residues on Kelch-Like ECH-Associated Protein 1 (KEAP1), the protein that normally holds Nrf2 down and tags it for destruction. R
When KEAP1 is modified, it releases Nrf2, which moves into the nucleus and switches on a battery of antioxidant and anti-inflammatory genes, including those for glutathione synthesis and heme oxygenase-1. R
This is the same target hit by sulforaphane and dimethyl fumarate, which is why itaconate is often described as an endogenous Nrf2 activator.
In Jacob's framework these are the Antioxidant Response Enzymes (AREs) that drive hormesis, and itaconate is one of the body's internal ways of turning them on.
NLRP3 inflammasome inhibition
The third mechanism is inhibition of the NLRP3 inflammasome, the protein complex that processes and releases IL-1 beta and IL-18 and triggers the inflammatory cell death called pyroptosis.
Itaconate alkylates cysteine 548 on NLRP3, which blocks it from interacting with NEK7 and prevents the inflammasome from assembling. R
This effect was shown to dampen IL-1 beta release in cells from patients with cryopyrin-associated periodic syndrome and to reduce inflammation in a model of gout-like urate crystal inflammation. R
The NLRP3 inflammasome is central to a long list of chronic inflammatory and metabolic diseases, so a metabolite that selectively quiets it is mechanistically significant.
The IkBzeta and ATF3 axis
The fourth mechanism is induction of Activating Transcription Factor 3 (ATF3), which then suppresses a pro-inflammatory factor called IkBzeta. R
Through electrophilic stress and reaction with glutathione, itaconate and its derivatives drive ATF3 to shut down a specific secondary wave of inflammatory genes, including IL-6, while sparing the early TNF response. R
In a mouse model of psoriasis, an itaconate derivative reduced the IL-17 and IkBzeta-driven skin pathology, which links this axis to real disease. R
A fifth and newer mechanism is worth flagging: itaconate analogs also alkylate Stimulator of Interferon Genes (STING), the sensor that drives type I interferon, which adds a brake on the interferon arm of inflammation as well. R
The Antimicrobial Angle
Itaconate is not only an off-switch for inflammation.
Before its anti-inflammatory role was understood, it was identified as a direct antimicrobial weapon. R
It inhibits a bacterial enzyme called isocitrate lyase, the key enzyme of the glyoxylate shunt. R
The glyoxylate shunt is a metabolic bypass that lets bacteria such as Mycobacterium tuberculosis and Salmonella survive on fat-derived carbon when sugar is scarce, which is exactly the situation they face hiding inside a macrophage. R
By forming a covalent adduct with the catalytic site of isocitrate lyase, itaconate starves these intracellular bacteria of a survival pathway. R
This is a clean example of an immunometabolite doing double duty, poisoning the pathogen's metabolism and calming the host's inflammation at the same time.
It also reframes how to think about persistent intracellular infection.
In Jacob's terrain-leaning view, the goal is to remove the toxic environment that drives pleomorphism rather than to brute-force kill, and a metabolite that simultaneously starves a pathogen and restores immune order fits that philosophy better than a blunt antibiotic.
Itaconate And Chronic Illness
This is where itaconate intersects directly with the Junction Dysfunction (JD) framework.
Itaconate is made by activated, M1-phenotype macrophages, the same cells that get stuck in chronic illness.
In Jacob's model of Micro-Sepsis (MSS), the sub-lethal chronic sepsis that he coined to describe the immune state behind post-viral illness, macrophages are driven by constant LPS exposure into a state of endotoxin tolerance and exhaustion where they are inflammatory and immunosuppressive at the same time.
You can read the full mechanism in the JD chapters on Micro-Sepsis and why the innate immune system gets stuck.
Itaconate is the body's attempt to apply its own brake inside this loop, by inhibiting SDH, activating Nrf2, and blocking NLRP3.
Across preclinical models, itaconate and its derivatives are protective in sepsis, viral infection, gout, psoriasis, ischemia-reperfusion injury, and pulmonary fibrosis. R
In viral infection the picture is genuinely mixed, and this is worth being honest about.
The itaconate derivative 4-octyl itaconate restricts replication of SARS-CoV-2 and other viruses by restoring Nrf2 signaling that the virus tries to suppress, which is a clear antiviral benefit. R
But because itaconate is also immunosuppressive, sustained high levels can blunt the antiviral interferon response and, in some settings, actually favor pathogen persistence. R
There is a big MAYBE here, and it maps onto the same tension Jacob describes in MSS, where the body's own anti-inflammatory and immunosuppressive responses, deployed for protection, can become part of why the immune system stays stuck and why latent infections re-express.
On the antibody question, this also fits Jacob's framing that autoantibodies are cleanup signatures of an inflammatory cascade rather than an attack, since the upstream driver is the stuck inflammatory state that itaconate is trying, and often failing, to resolve.
The Research Tools And Why There Is No Clean Supplement
Almost everything exciting about itaconate has been shown using two laboratory analogs, not itaconate itself.
The reason is simple chemistry.
Itaconate is charged and does not cross cell membranes well, so feeding it to cells or to a person does not reliably get it inside the cells where it works.
To get around this, researchers use cell-permeable esters that slip through the membrane and then convert, or are presumed to convert, to itaconate or an itaconate-like species inside the cell.
- 4-Octyl Itaconate (4-OI): the most widely used tool, responsible for most of the Nrf2, NLRP3, STING, and antiviral data above R
- Dimethyl Itaconate (DI): the tool used in the original ATF3 and IkBzeta work and the psoriasis model R

Here is the important caveat that the marketing around this molecule tends to skip.
Neither 4-OI nor DI is simply itaconate, and there is real debate about how much of their effect comes from genuine itaconate biology versus their own independent electrophilic activity.
DI in particular barely raises intracellular itaconate and may act largely through its own chemistry.
These are research compounds, not vetted human supplements, and they have not been through the safety and dosing studies that would justify taking them.
There is no clean, proven oral itaconate supplement on the market that has been shown to safely raise the active metabolite inside human immune cells.
Anyone selling you "itaconate" as a finished anti-inflammatory pill is ahead of the evidence.
The honest, practical move is to support the pathway that makes itaconate and to activate the same downstream targets through better-studied means.
How To Support The Endogenous Pathway
The realistic strategy is twofold: keep the immune signal that turns on ACOD1 working, and hit the same downstream targets (especially Nrf2) with tools that actually have human data.
These overlap heavily with the glycocalyx and mitochondrial recovery logic in the JD guide, so this is not a separate protocol so much as the same one viewed through the itaconate lens.
1. Activate Nrf2 with sulforaphane
Sulforaphane from broccoli sprouts is the best-studied natural Nrf2 activator and hits the same KEAP1 and Nrf2 axis that itaconate does. R
See the full writeup on broccoli sprouts and sulforaphane for sourcing and preparation.
2. Restore the ability to use Nrf2 with DJ-1 support
Nrf2 activators do nothing if Nrf2 cannot translocate into the nucleus, which requires the protein DJ-1.
This is why some people feel worse, not better, on Nrf2 activators, a problem covered in why NRF2 activation can make you more sick and in the deep dive on reviving NRF2 through DJ-1.
Supports redox recycling alongside Nrf2 activation.
3. Use DIM as an additional Nrf2 lever
DIM:
Diindolylmethane is one of Jacob's preferred Nrf2 activators and immunomodulators and shares the antiviral, Nrf2-restoring logic seen with 4-OI in the SARS-CoV-2 work. R
4. Lower the LPS load that keeps macrophages inflamed
If chronic endotoxin exposure is what keeps your macrophages locked in the inflamed state, reducing that load is upstream of everything else.
This is the entire subject of how to inhibit LPS and endotoxins.
5. Feed the gut to lower inflammatory tone
Short chain fatty acids dampen NF-kB and TLR4 signaling and support the same anti-inflammatory macrophage shift, covered in the post on short chain fatty acids.
6. Use fasting and ketosis as a metabolic lever
Beta-hydroxybutyrate is itself an endogenous NLRP3 inhibitor, which means a fasting or ketogenic window applies a second brake on the same inflammasome itaconate targets.
This pairs with the glycocalyx repair window that opens during fasting, described in the JD chapter on healing the glycocalyx.
What To Stay Away From
Avoid these in the itaconate context (not an exclusive list):
- Blind Nrf2 stacking without addressing DJ-1 and redox capacity first, because pushing Nrf2 in someone who cannot use it can worsen sensitivities R
- Chronic high-dose antioxidant megadosing, which can blunt the hormetic electrophilic signaling that itaconate and sulforaphane depend on
- Research-chemical "itaconate" or 4-OI powders sold as supplements, which have no human safety or dosing data R
- Unmanaged high blood sugar, which sustains the inflammatory macrophage state and works against the entire pathway
Testing
Itaconate itself is not yet a routine clinical blood test, so the practical approach is to measure the upstream drivers and the downstream inflammatory output it is meant to control.
Blood And Urine Markers
Succinate and organic acids are worth tracking, because elevated succinate and related Krebs cycle intermediates reflect the metabolic state that itaconate normally counterbalances, and these show up on organic acid testing. R
Inflammatory cytokines (IL-1 beta, IL-6, TNF-alpha, hsCRP) are the downstream outputs of the SDH, NLRP3, and IkBzeta pathways itaconate suppresses. R
Functional Lab Panels
I use the Cellular Zoomer (Vibrant Wellness) to assess organic acids, mitochondrial function, and oxidative stress, which together reflect the metabolic environment around the itaconate pathway.
I use the Cardio Zoomer (Vibrant Wellness) to assess inflammatory and endothelial markers when the question is systemic inflammatory tone.
For autoantibody and mast cell activity in a chronically inflamed picture, the Immune Zoomer (Vibrant Wellness) maps the adaptive immune cleanup response sitting downstream of stuck innate inflammation.
If the underlying driver is endotoxin and gut permeability, the Gut Zoomer (Vibrant Wellness) assesses microbiome, zonulin, and the LPS sources that keep macrophages activated.
For a focused post-viral workup, the Long COVID bundle combines the cardio, toxin, gut, cellular, and viral panels most relevant to a stuck inflammatory state.
If you want help interpreting these in the context of MSS and Junction Dysfunction, this is the kind of pattern I work through on a consultation.
Mechanisms Of Action
Simple:
- Your immune cells make itaconate from a Krebs cycle molecule to calm their own inflammation once a threat is handled.
- It works by chemically tagging a handful of key proteins, which turns on your antioxidant defenses and turns off several inflammation switches at once.
- It also poisons a metabolic shortcut that bacteria hiding inside your cells need to survive.
Advanced:
- ACOD1 / IRG1 induction: Upon TLR4 ligation by LPS, myeloid cells strongly upregulate ACOD1, which decarboxylates cis-aconitate to itaconate, creating a Krebs cycle "break" that diverts carbon away from oxidative phosphorylation. R
- SDH inhibition: Itaconate competitively inhibits succinate dehydrogenase (complex II), lowering succinate-driven HIF-1 alpha stabilization and IL-1 beta, and reducing reverse electron transport ROS. R
- KEAP1 alkylation: Itaconate alkylates KEAP1 cysteines, freeing Nrf2 (NFE2L2) for nuclear translocation and transcription of antioxidant and anti-inflammatory genes including glutathione and HO-1. R
- NLRP3 modification: Itaconate dicarboxypropylates cysteine 548 of NLRP3, blocking the NLRP3-NEK7 interaction and inflammasome assembly, reducing caspase-1 activation and IL-1 beta release. R
- ATF3 / IkBzeta axis: Through electrophilic stress and glutathione depletion, itaconate and its derivatives induce ATF3, which suppresses IkBzeta and a secondary wave of inflammatory transcription including IL-6, sparing early TNF. R
- STING alkylation: Itaconate derivatives alkylate STING and block its palmitoylation and oligomerization, dampening type I interferon output. R
- Isocitrate lyase inhibition: Itaconate covalently inhibits bacterial isocitrate lyase, the rate-limiting enzyme of the glyoxylate shunt, starving intracellular pathogens that depend on fatty-acid-derived carbon. R
Genetics
ACOD1 (IRG1)
ACOD1 encodes aconitate decarboxylase 1, the single enzyme responsible for producing itaconate from cis-aconitate.
Loss or reduced induction of ACOD1 removes the body's ability to deploy this brake, and Acod1-deficient animals show exaggerated inflammation and worse outcomes in sepsis and infection models. R
ACOD1 expression is strongly induced by TLR signaling rather than fixed by a single common variant, so functional output depends heavily on the inflammatory environment, not just genotype.
NFE2L2
NFE2L2 encodes Nrf2, the transcription factor that itaconate activates by modifying KEAP1.
Variants that lower baseline Nrf2 activity blunt the antioxidant payoff of itaconate and of Nrf2 activators like sulforaphane, while variants that raise activity can shift the balance. R
In the JD population, the more practical genetic question is whether the downstream redox machinery works at all, which points to SOD2 at rs4880 and the sulfation genes (SUOX, CBS) on Jacob's priority SNP list rather than to NFE2L2 alone.
Downstream redox capacity
A normal Nrf2 response still requires DJ-1 (PARK7) to allow Nrf2 nuclear translocation, so impaired DJ-1 function can leave the itaconate-Nrf2 axis switched on at the top but non-functional downstream. R
This is the genetic and functional reason some people react badly to Nrf2 activation, covered in the DJ-1 post linked above.
More Research
Citraconate, a structural isomer of itaconate, is actually a more potent inhibitor of certain targets and competitively inhibits ACOD1 itself, which complicates the simple picture of "more itaconate is better" and is an active area of investigation. R
Dimethyl fumarate, the multiple sclerosis and psoriasis drug sold as Tecfidera, works through the same electrophilic Nrf2-activating chemistry as the itaconate analogs and shares antiviral activity against SARS-CoV-2 in cell models, which is a useful real-world proof of concept for the class even though it is a prescription drug, not an itaconate. R
For biomarker tracking around this pathway I use the Cellular Zoomer to follow organic acids and oxidative stress over time, since itaconate itself is not yet a standard clinical assay.
The biggest open question is whether itaconate is net protective or net harmful in any given chronic infection, because the same immunosuppression that calms inflammation can also let pathogens persist, and the honest answer is that it depends on timing, dose, and the specific infection. R
The translational gap is real: nearly all human-relevant data comes from 4-octyl itaconate and dimethyl itaconate, both of which have independent chemistry of their own, so claims made about "itaconate" based on these tools should be read with that caveat in mind. R
If you want to go deeper on how this fits the stuck-macrophage state behind post-viral illness, the Junction Dysfunction guide and the long COVID protocol are the next reads.
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Quercetin
500mg 2x/day
SPM Active (Pro-resolving Mediators)
2 softgels/day
Curcumin (Liposomal)
500mg 2x/day