Xanthine And Xanthine Oxidase: The Molecule In Your Coffee, The Enzyme Behind Gout, And Why Both Matter For Your Health
By Jacob Gordon, INHC, FMT-CEvery cup of coffee you drink contains methylated derivatives of xanthine.
Every episode of gout involves a failure to clear xanthine fast enough.
And every ischemia-reperfusion injury, whether from a heart attack, stroke, or surgical clamp, involves xanthine oxidase generating a burst of superoxide that tears through recovering tissue.
In this post, we will cover what xanthine is and where it sits in purine metabolism, the full methylxanthine family in food and pharmacology, how xanthine oxidase works at the enzyme level, the XDH-to-XO conversion that drives ischemic injury, uric acid homeostasis and why humans lost uricase, gout and NLRP3 inflammasome activation, the fructose-uric acid connection, cardiovascular and metabolic disease implications, and what actually moves the needle therapeutically.

What Xanthine Is: The Purine Scaffold
Xanthine (3,7-dihydro-1H-purine-2,6-dione) is a purine base, meaning it shares the bicyclic purine ring structure: a six-membered pyrimidine ring fused to a five-membered imidazole ring.
It sits at a junction in purine metabolism: xanthine is produced from multiple upstream purines, and it is the immediate precursor to uric acid.
Xanthine is formed from three distinct upstream reactions in purine catabolism:
- From hypoxanthine by xanthine oxidoreductase (the dominant route in humans)
- From guanine by guanine deaminase (GDA)
- From xanthosine by purine nucleoside phosphorylase (PNP)
Xanthine is then irreversibly converted to uric acid by xanthine oxidase, which is the final step in the human purine degradation pathway.
Xanthine itself is not a central metabolic fuel or signaling molecule in the way that ATP or AMP are.
Its biological significance comes from two directions: the methylated derivatives that plants synthesize from it (caffeine, theophylline, theobromine), and the enzyme that processes it (xanthine oxidoreductase) whose activity determines uric acid production and reactive oxygen species generation.
The Methylxanthines In Your Food: Caffeine, Theophylline, Theobromine
Methylxanthines are methylated derivatives of xanthine produced by a small number of plant species, apparently as a defense mechanism against insects and pathogens. R
The three primary naturally occurring methylxanthines:
Caffeine (1,3,7-trimethylxanthine):
Three methyl groups at positions 1, 3, and 7 of the xanthine ring. R
The most widely consumed psychoactive substance in the world. R
Found in coffee (Coffea arabica, Coffea robusta), tea (Camellia sinensis), guarana, kola nut, and yerba mate.
Typical coffee cup: 70-150 mg caffeine.
Half-life in healthy adults: approximately 2.5-5 hours, highly variable depending on CYP1A2 activity and smoking status. R
Theophylline (1,3-dimethylxanthine):
Two methyl groups at positions 1 and 3. R
Found in tea at low concentrations alongside caffeine; approximately 4% of hepatic caffeine demethylation produces theophylline. R
Used therapeutically as a bronchodilator for asthma and COPD.
Has higher potency than caffeine at adenosine receptors (Ki theophylline 6.7 µM vs caffeine 9.5 µM at A2A) despite lower subjective CNS stimulant effect. R
Narrow therapeutic index in clinical use; serum levels must be monitored.
Theobromine (3,7-dimethylxanthine):
Two methyl groups at positions 3 and 7.
Dominant methylxanthine in cocoa (Theobroma cacao) and therefore dark chocolate. R
Found in small quantities in tea; approximately 11% of hepatic caffeine demethylation produces theobromine. R
Much weaker adenosine receptor antagonist than caffeine or theophylline. R
More prominent as a vasodilator and cardiac stimulant than as a CNS stimulant.
The "theobromine poisoning" in dogs results from their inability to metabolize it as rapidly as humans can.
Paraxanthine (1,7-dimethylxanthine):
The primary hepatic metabolite of caffeine in humans, produced by CYP1A2-mediated N-3 demethylation. R
Approximately 80-90% of caffeine is metabolized to paraxanthine via this route, exclusively by CYP1A2. R
Roughly equipotent to caffeine at adenosine receptors (Ki 21 µM at A1, 32 µM at A2A). R
Increasingly studied independently as a potentially cleaner stimulant with some of caffeine's benefits but fewer downsides.
Which plants produce which:
- Coffee: caffeine dominant, trace theophylline
- Tea: caffeine plus significant theophylline and theobromine at low levels
- Cocoa/dark chocolate: theobromine dominant, small amounts of caffeine
- Guarana: caffeine at approximately twice the concentration of coffee by weight
- Yerba mate: caffeine, theobromine, and theophylline
How Methylxanthines Work: Adenosine Receptor Antagonism And PDE Inhibition
Methylxanthines produce their physiological effects through two primary mechanisms:
1. Adenosine receptor antagonism:
Adenosine (adenine + ribose, without a phosphate group) is produced continuously by cellular metabolism, especially during periods of high activity. R
Adenosine acts as a neuromodulator that signals metabolic stress and promotes sleep, reduces neuronal firing rates, dilates blood vessels in active tissue, and generally acts as a brake on excitatory signaling.
Methylxanthines are competitive nonselective antagonists at all four adenosine receptor subtypes (A1, A2A, A2B, A3), blocking adenosine's access to its receptors without activating them. R
The receptor-specific effects:
- A1 antagonism: disinhibits hippocampal glutamate release, contributing to memory enhancement; causes diuresis; causes cardiac tachycardia R
- A2A antagonism: the primary mechanism of alertness; A2A receptors on striatal neurons normally suppress dopaminergic signaling when adenosine is present; blocking A2A releases this inhibition and increases dopamine signaling, producing wakefulness and alertness R
- A2B antagonism: contributes to bronchodilation (relevant for theophylline's therapeutic use in asthma)
- A3 antagonism: less characterized; may contribute to cardiovascular and immune effects
Caffeine and theophylline are equipotent or theophylline is slightly more potent at adenosine receptors, but the subjective stimulant effect of caffeine is greater in most people, likely due to differences in CNS distribution, metabolism, and receptor selectivity profiles across tissues. R
Theobromine is a weak adenosine receptor antagonist; its effects are primarily cardiovascular and bronchopulmonary, not predominantly CNS stimulant. R
2. Phosphodiesterase (PDE) inhibition:
Methylxanthines nonselectively inhibit phosphodiesterase enzymes (PDEs), which break down cyclic AMP (cAMP) and cyclic GMP (cGMP). R
PDE inhibition raises intracellular cAMP, which:
- Activates protein kinase A (PKA)
- Inhibits TNF-alpha synthesis
- Suppresses leukotriene synthesis
- Reduces inflammation in macrophages and mast cells
- Increases bronchial smooth muscle relaxation (theophylline's bronchodilator mechanism)
At physiological doses from dietary caffeine, adenosine receptor antagonism is the dominant mechanism. R
PDE inhibition becomes more significant at higher therapeutic concentrations, particularly with theophylline. R
Purine Catabolism: The Full Pathway To Uric Acid
Understanding uric acid production requires understanding where all the substrate comes from.
The purine nucleotides (ATP, ADP, AMP, GTP, GDP, GMP) are degraded through a sequential series of reactions: R
From the adenine nucleotides:
ATP → ADP → AMP → IMP (inosine monophosphate) via adenosine deaminase
IMP → inosine via 5'-nucleotidase
Inosine → hypoxanthine + ribose-1-phosphate via purine nucleoside phosphorylase (PNP)
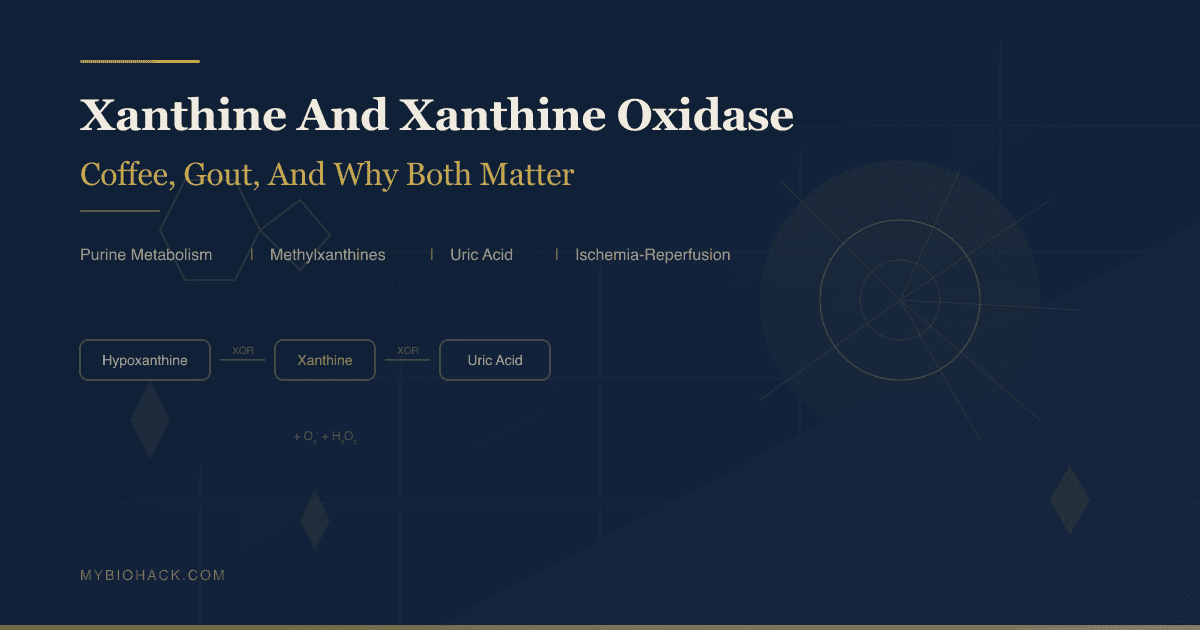
Hypoxanthine → xanthine → uric acid (both steps catalyzed by xanthine oxidoreductase)
From the guanine nucleotides:
GTP → GDP → GMP → guanosine (via 5'-nucleotidase)
Guanosine → guanine + ribose-1-phosphate (via PNP)
Guanine → xanthine (via guanine deaminase/GDA) R
Xanthine → uric acid (via xanthine oxidoreductase)
The salvage pathway runs in the opposite direction and recycles purines back to nucleotides: R
Hypoxanthine + PRPP → IMP (via HGPRT, hypoxanthine-guanine phosphoribosyltransferase)
HGPRT is critical for preventing uric acid accumulation.
When HGPRT activity is high, hypoxanthine is recycled to IMP without consuming ATP, making the salvage pathway energetically efficient. R
Lesch-Nyhan syndrome, a rare X-linked disorder, results from complete HGPRT deficiency: without the salvage pathway, all hypoxanthine goes to uric acid, causing severe gout, kidney stones, and neurological dysfunction including self-mutilation. R
De novo purine synthesis is the other major upstream driver of uric acid:
The liver synthesizes purines from scratch starting with ribose-5-phosphate (from the pentose phosphate pathway) through a 10-step series of reactions to produce IMP. R
This pathway is activated by:
- Elevated PRPP (phosphoribosyl pyrophosphate) availability R
- Fructose metabolism (see the fructose section below)
- ATP depletion (the cell responds by making more purines)
Sources of purine substrate for degradation:
- Cell turnover (DNA and RNA breakdown from dying cells) R
- Dietary purines (meat, organ meats, seafood, beer)
- De novo synthesis (especially when stimulated by fructose or ischemia)
- Nucleotide catabolism during intense exercise (ATP → ADP → AMP → xanthine/uric acid in muscle) R
Xanthine Oxidoreductase: The Enzyme In Detail
Xanthine oxidoreductase (XOR) is the enzyme that catalyzes both:
- Hypoxanthine → xanthine
- Xanthine → uric acid
Both reactions are oxidative hydroxylations at the C-8 and C-2 positions of the purine ring, respectively.
Molecular structure:
XOR is a homodimer with a molecular mass of approximately 290 kDa.
Each monomer contains three redox-active components arranged in an electron transfer chain: R
- Molybdopterin (Mo-pterin) cofactor: The active site where substrate binds and is oxidized. Molybdenum cycles between Mo(VI) and Mo(IV) oxidation states as it accepts electrons from the substrate. Xanthine binds at this site, is hydroxylated, and uric acid is released.
- Two iron-sulfur clusters (2Fe-2S): Serve as relay stations for electron transfer from the Mo center to the FAD cofactor.
- FAD (flavin adenine dinucleotide): The final electron acceptor from the iron-sulfur clusters. FAD passes electrons to the terminal electron acceptor, which differs between the two forms of the enzyme.
The two interconvertible forms:
Xanthine Dehydrogenase (XDH):
The predominant form in most tissues under normal physiological conditions. R
Uses NAD+ as the terminal electron acceptor.
When NAD+ accepts electrons: NAD+ → NADH.
This is a "clean" transfer that does not generate ROS.
XDH is the form the gene encodes; XOR is transcribed and translated as XDH. R
Xanthine Oxidase (XO):
Uses molecular oxygen (O2) as the terminal electron acceptor.
When O2 accepts electrons:
- Univalent reduction: O2 + e- → superoxide (O2-), which is the dominant product
- Divalent reduction: O2 + 2e- → hydrogen peroxide (H2O2)
This means XO, unlike XDH, generates reactive oxygen species as a direct byproduct of purine catabolism. R
XDH can be converted to XO by:
- Reversible sulfhydryl oxidation: oxidation of cysteine residues at the FAD domain changes the electron affinity such that NAD+ can no longer compete effectively with O2; this is reversible by reducing agents R
- Irreversible proteolytic cleavage: specific proteases cleave the protein, causing a conformational change that permanently locks in the XO form R
Tissue distribution:
XOR is expressed most highly in the liver (the major site of purine catabolism) and the intestine (an important but underappreciated site). R
Lower but clinically relevant expression in: vascular endothelium, heart, lung, kidney, brain, skeletal muscle. R
Plasma XOR activity reflects primarily hepatic and intestinal release of the enzyme into circulation.
The XDH-To-XO Switch: How Ischemia Arms The Enzyme
This is one of the most elegantly dangerous mechanisms in human biology.
Under normal conditions, most tissue XOR exists in the XDH form.
XDH produces NADH, not superoxide.
The tissue accumulates minimal oxidative stress from purine catabolism.
During ischemia (absence of blood flow, oxygen deprivation):
- ATP is rapidly consumed as cells try to maintain function without oxidative phosphorylation.
- ATP → ADP → AMP → hypoxanthine + xanthine (the purine catabolism cascade runs rapidly because the substrate, ATP, is being depleted and there is no energy to resynthesize it).
- Intracellular hypoxanthine and xanthine accumulate massively because there is no product (uric acid) that can be cleared without blood flow.
- Simultaneously, hypoxia activates proteases that irreversibly convert XDH to XO in the ischemic tissue. R
- The converted enzyme (XO) is now primed and loaded with massive substrate (hypoxanthine/xanthine) but cannot act because there is no oxygen to accept the electrons.
At reperfusion (blood flow and oxygen restored):
The oxygen-starved tissue is suddenly flooded with both O2 and massive xanthine/hypoxanthine substrate simultaneously.
XO, now present in large quantities and with high enzyme kinetics, catalyzes the oxidation of all accumulated substrate rapidly.
The result is a burst of superoxide and H2O2 production far exceeding anything that occurs under normal homeostatic conditions.
This superoxide burst:
- Reacts with nitric oxide (NO) to form peroxynitrite (ONOO-), which nitrosylates proteins and DNA R
- Activates the NLRP3 inflammasome R
- Triggers NF-kB and pro-inflammatory cytokine release (TNF-alpha, IL-1beta, IL-6) R
- Activates neutrophils to adhere to the endothelium and generate additional ROS via their own NADPH oxidase
- Damages mitochondria and initiates apoptosis in previously viable cells
This is ischemia-reperfusion injury, and xanthine oxidase is one of its primary molecular executioners. R
The XO burst also explains why the damage from restoring blood flow after a heart attack or stroke often exceeds the damage from the ischemia itself. R
Uric Acid: Antioxidant Or Pro-Oxidant?
Uric acid's biology is genuinely paradoxical, and the paradox matters clinically.
The antioxidant case:
Uric acid is the primary antioxidant in human plasma. R
It scavenges peroxynitrite, hydroxyl radical, and singlet oxygen. R
It chelates iron, reducing iron's ability to catalyze Fenton reactions (Fe2+ + H2O2 → Fe3+ + OH- + OH•).
Uric acid is estimated to account for up to 60% of total plasma antioxidant capacity. R
When uricase was lost in the primate lineage (more on this below), uric acid levels rose substantially compared to other mammals.
This elevation may have provided an evolutionary advantage in terms of antioxidant protection, particularly during periods of oxidative stress.
The pro-oxidant case:
Inside cells and in inflamed tissues, uric acid behaves differently than in plasma. R
At high concentrations, uric acid crystals trigger the NLRP3 inflammasome, a major driver of sterile inflammation. R
Soluble uric acid activates NADPH oxidase in macrophages and endothelial cells, generating intracellular superoxide. R
Soluble uric acid inhibits endothelial nitric oxide synthase (eNOS), reducing NO bioavailability and impairing vasodilation. R
Elevated uric acid activates the renin-angiotensin system and promotes oxidative stress in the kidney, driving hypertension. R
In the liver, elevated uric acid from fructose metabolism activates XOR activity in a feed-forward loop, generating more ROS. R
The resolution of the paradox:
The antioxidant versus pro-oxidant behavior of uric acid depends on:
- Concentration: plasma concentrations within normal range are largely antioxidant; above 6.8 mg/dL, pro-inflammatory effects dominate
- Location: extracellular plasma uric acid is antioxidant; intracellular uric acid is pro-oxidant
- Physical state: soluble uric acid versus crystalline monosodium urate (MSU), where crystals are specifically and potently inflammogenic
- Context: acute elevations (exercise, oxidative stress) may be protective; chronic hyperuricemia drives disease R
Why Humans Kept Uric Acid: The Uricase Loss
Most mammals convert uric acid to allantoin via uricase (urate oxidase), a peroxisomal enzyme.
Allantoin is far more water-soluble than uric acid, is non-inflammatory, and is easily excreted.
Mammals with intact uricase maintain serum uric acid levels of approximately 0.5-1.5 mg/dL.
In higher primates including humans, the uricase gene was inactivated approximately 15-20 million years ago through two independent mutational events, making us effectively uricase-deficient by evolutionary design. R
As a result, normal human serum uric acid levels (3-7 mg/dL) are roughly 5-10 times higher than in other mammals with intact uricase. R
Why did natural selection allow this to persist?
Several hypotheses have been proposed: R
- Antioxidant advantage: Higher uric acid provided an antioxidant benefit during a period of dietary transition when ancestral hominids had reduced vitamin C intake (due to loss of gulonolactone oxidase even earlier in primate evolution). Uric acid may have partially compensated for reduced ascorbate. R
- Blood pressure and sodium conservation: Higher uric acid enhances sensitivity to sodium, potentially helping blood pressure maintenance during evolutionary periods of salt scarcity. R
- Fructose absorption efficiency: Higher uric acid and lower uricase activity allows fructose to generate more AMP-driven IMP accumulation, driving de novo fat synthesis from fructose, potentially an advantage during periods when fructose from fruit was a dense caloric opportunity.
Whatever the ancestral advantage, the consequence is clear: humans are uniquely vulnerable to hyperuricemia, gout, and the inflammatory sequelae of urate crystal deposition in a way that most mammals are not. R
Hyperuricemia: When The System Breaks Down
Hyperuricemia is defined as serum uric acid above 6.8 mg/dL (the physiological solubility threshold of monosodium urate at body temperature) in men and above 6.0 mg/dL in women (women have lower baseline levels, partly due to estrogen's uricosuric effect). R
Above the saturation threshold, uric acid begins crystallizing into monosodium urate (MSU) crystals, particularly in cooler peripheral tissues: toe joints, ankle joints, the pinna of the ear, and the kidneys. R
Two mechanisms drive hyperuricemia:
Overproduction (~10% of cases): R
- High purine diet (organ meats, red meat, shellfish, anchovies)
- Elevated cell turnover (hemolytic anemia, tumor lysis syndrome, myeloproliferative disorders) R
- Fructose overconsumption (via ATP depletion and de novo synthesis)
- Alcohol (beer especially contains purines; alcohol also impairs renal urate excretion and drives de novo synthesis)
- Inborn errors of purine metabolism (HGPRT deficiency, PRPP synthetase superactivity)
Underexcretion (~90% of cases, often the dominant mechanism): R
The kidney filters uric acid freely at the glomerulus but reabsorbs approximately 90% of it in the proximal tubule. R
Key renal transporters: R
- URAT1 (SLC22A12): apical reabsorption transporter, dominant driver of tubular urate recapture; target of uricosuric drugs
- GLUT9 (SLC2A9): basolateral transporter moving uric acid from tubule cell to blood; also a fructose transporter (relevant to the fructose connection) R
- ABCG2: apical secretory transporter; loss-of-function variants significantly impair intestinal and renal urate excretion and are a major genetic determinant of gout risk R
- OAT1/OAT3 (SLC22A6/8): basolateral uptake transporters; organic anion exchangers
Insulin resistance increases URAT1 and GLUT9 expression, reducing net renal urate secretion. R
Estrogen suppresses URAT1 and GLUT9 and upregulates ABCG2, explaining why premenopausal women are substantially protected from gout, and why incidence equalizes after menopause. R
Hepatic XOR activity in females is approximately 40% lower than males, with ovariectomy restoring it to male levels. R
Gout: Crystals, NLRP3, And The Inflammatory Cascade
Gout is the inflammatory disease caused by MSU crystal deposition in joints and soft tissues.
It is the most common form of inflammatory arthritis in adults.
The acute gout attack mechanism:
Step 1: Crystal formation
When serum urate exceeds 6.8 mg/dL at physiological temperature (and lower concentrations in cooler peripheral joints such as the first metatarsophalangeal joint, the classic site of podagra), monosodium urate begins crystallizing. R
Crystals accumulate preferentially in avascular cartilage, synovial tissue, and the renal collecting ducts (where they cause kidney stones and, in severe cases, urate nephropathy). R
Step 2: Crystal recognition by macrophages
MSU crystals are phagocytized by synovial macrophages and other innate immune cells in the affected joint.
However, MSU crystals are physically too large to be fully degraded by the phagolysosome.
The crystal damages the phagolysosomal membrane (a process called lysosomal rupture), releasing cathepsins and other lysosomal contents into the cytoplasm. R
Step 3: NLRP3 inflammasome activation
MSU crystal-induced lysosomal damage, combined with concurrent xanthine oxidase-generated ROS, activates the NLRP3 inflammasome, a cytoplasmic multiprotein complex that serves as a sensor for cellular damage. R
NLRP3 activation assembles ASC (apoptosis-associated speck-like protein) and recruits caspase-1.
Active caspase-1 cleaves pro-IL-1beta to mature IL-1beta and pro-IL-18 to IL-18.
IL-1beta is released and acts as the primary driver of the acute inflammatory cascade. R
Step 4: Neutrophil recruitment and the pain of gout
IL-1beta drives neutrophil recruitment into the joint in massive numbers. R
Neutrophils also try to phagocytize MSU crystals, also rupture their phagolysosomes, and also release their contents into the joint space, amplifying the inflammation.
The NET (neutrophil extracellular trap) formation that follows, combined with oxidative burst from neutrophil NADPH oxidase, creates the intensely painful, swollen, erythematous joint of acute gout.
Untreated attacks typically resolve spontaneously in 7-14 days as the inflammatory cascade self-limits, but joint damage accumulates with recurrent attacks. R
Tophi are deposits of urate crystals surrounded by macrophage-driven granulomatous inflammation, forming over years in joints, tendons, and soft tissues. R
XOR's direct role in inflammasome activation (beyond crystal formation):
XOR within activated macrophages itself generates ROS that contribute to NLRP3 activation, independent of crystal formation. R
XOR inhibition reduces IL-1beta release even when urate crystals are present, suggesting XO-derived ROS are required co-signals for full inflammasome assembly. R
This is one reason why XOR inhibitors may have anti-inflammatory benefits beyond their urate-lowering effects. R
The Fructose-Uric Acid Connection
The fructose-to-uric-acid pathway is one of the most important and underappreciated connections in metabolic disease.
Why fructose spikes uric acid:
Glucose and fructose enter cells differently.
Glucose phosphorylation (via hexokinase/glucokinase) is tightly regulated by product inhibition and is limited by the Km of the enzyme.
Fructose phosphorylation (via fructokinase/KHK) is not product-inhibited and is essentially irreversible at physiological concentrations. R
When fructose enters the liver in large amounts:
- Fructokinase converts fructose to fructose-1-phosphate, consuming ATP rapidly R
- ATP depletion creates AMP accumulation
- AMP is then catabolized: AMP → IMP → inosine → hypoxanthine → xanthine → uric acid R
- Simultaneously, the AMP accumulation and ATP depletion stimulate de novo purine synthesis via PRPP synthetase upregulation, creating even more purine substrate R
- Fructose metabolism also increases hepatic XOR transcription and activity directly, accelerating the conversion of hypoxanthine to uric acid R
This mechanism is why a single large fructose load can raise serum uric acid within hours, and why chronic high fructose consumption (from high fructose corn syrup, table sugar, and concentrated fruit juices) is a major driver of hyperuricemia. R
Critically, this effect is calorie-independent in some studies: high-fructose diets raise uric acid through mechanisms independent of weight gain and energy excess. R
Isocaloric fructose substituted for other carbohydrates still elevates uric acid.
The sugar-to-gout pathway: sugar consumption rose dramatically in Western countries over the 20th century, and gout prevalence tracked that rise closely. R
Beer is uniquely gout-promoting: it contains purines (from yeast), alcohol (impairs urate excretion), and promotes elevated uric acid through multiple mechanisms simultaneously. R
Xanthine Oxidase In Cardiovascular And Metabolic Disease
XOR activity links purine metabolism to cardiovascular and metabolic disease through several mechanisms:
Endothelial dysfunction:
XO-derived superoxide reacts with nitric oxide (NO) at near-diffusion-limited rates to produce peroxynitrite (ONOO-). R
Peroxynitrite is more reactive than either superoxide or NO separately and causes:
- Lipid peroxidation
- Protein nitrosylation (including nitrosylation of eNOS itself, reducing further NO production)
- DNA damage
- Mitochondrial damage R
The net result is reduced NO bioavailability, which impairs endothelium-dependent vasodilation, promotes platelet activation, and contributes to atherosclerosis. R
Plasma XOR activity correlates with endothelial dysfunction markers in multiple clinical populations. R
Allopurinol treatment improves flow-mediated dilation (a measure of endothelial function) in hyperuricemic patients, with the improvement attributable to reduced XO-derived oxidative stress rather than the urate reduction per se. R
Hypertension:
Soluble uric acid activates the renin-angiotensin-aldosterone system (RAAS) in the kidney, reducing urinary sodium excretion. R
Uric acid also activates NADPH oxidase in smooth muscle cells, promoting vasoconstriction. R
In animal models, feeding fructose to rats induces hyperuricemia, hypertension, insulin resistance, and hypertriglyceridemia; all of these features are significantly blunted by allopurinol. R
Heart failure:
XOR activity is significantly elevated in the failing myocardium, particularly in the setting of ischemic heart failure. R
XO localizes within CD68-positive macrophages in failing heart tissue. R
Elevated plasma XOR activity predicts mortality in chronic heart failure. R
Intracoronary allopurinol in dilated cardiomyopathy patients reduced myocardial oxygen consumption and improved cardiac function, an effect not seen with uricosuric agents, suggesting the XO-derived ROS suppression (not the urate lowering) drove the cardiac benefit. R
Metabolic syndrome and insulin resistance:
Serum XOR activity correlates with the TG/HDL ratio, fasting glucose, fasting insulin, and HOMA-IR. R
XOR promotes adipogenesis and preadipocyte differentiation. R
Elevated uric acid drives insulin resistance by impairing insulin-stimulated glucose uptake in skeletal muscle via direct inhibition of Akt signaling. R
The metabolic syndrome cluster (central adiposity, hypertriglyceridemia, low HDL, hypertension, hyperglycemia) has a high prevalence of hyperuricemia that is not explained by gout risk alone. R
Ischemia-Reperfusion Injury: The XO Burst
Ischemia-reperfusion injury is one of the most clinically consequential contexts for xanthine oxidase biology.
During ischemia:
- ATP is depleted (ischemia stops oxidative phosphorylation)
- AMP accumulates, then is catabolized to hypoxanthine and xanthine
- XDH is proteolytically converted to XO
- Hypoxanthine and xanthine accumulate (no blood flow to clear them)
- XO is loaded with substrate but inactive (no O2)
At reperfusion:
- O2 is restored suddenly
- XO oxidizes all accumulated xanthine and hypoxanthine simultaneously
- Superoxide burst occurs in seconds to minutes
- Superoxide + NO → peroxynitrite
- NLRP3 inflammasome activation
- NF-kB activation → TNF-alpha, IL-1beta, IL-6 release
- Neutrophil adhesion and transmigration
- Tissue damage often exceeding the ischemic damage itself
XO has been documented as a major superoxide source in ischemia-reperfusion injury of: heart, brain (forebrain), skin, liver, kidney, skeletal muscle (tourniquet), and gastrointestinal mucosa. R
Circulating XO:
XO released from ischemic tissue enters the bloodstream.
Circulating XO binds to endothelial heparan sulfate and vascular cell adhesion molecules throughout the body, generating ROS at distant vascular sites.
This explains why local ischemia (e.g., limb tourniquet, liver ischemia) can produce systemic vascular and pulmonary injury at reperfusion.
XOR inhibitors given before reperfusion (pre-treatment) significantly reduce ischemic injury in multiple organ models.
In the clinical context, this has implications for cardiac surgery, organ transplantation, stroke management, and tourniquet use in orthopedic surgery.
Therapeutic Targeting: Allopurinol, Febuxostat, And Beyond
Allopurinol:
The first clinically approved XOR inhibitor, introduced in 1963.
A purine analogue (hypoxanthine analogue): allopurinol is itself a substrate for XOR, which oxidizes it to oxypurinol.
Oxypurinol binds tightly to the reduced form of the Mo-pterin active site, acting as a tight-binding inhibitor (not purely competitive).
The result: XOR is occupied by oxypurinol and cannot process hypoxanthine or xanthine.
Advantages: well-tolerated, once-daily dosing, effective regardless of cause of hyperuricemia.
Limitations:
- Allopurinol hypersensitivity syndrome (AHS): rare but potentially fatal (Stevens-Johnson syndrome, toxic epidermal necrolysis); strongly associated with HLA-B*5801 allele, particularly in Han Chinese, Thai, and Korean populations; genetic screening is now recommended before starting allopurinol in high-risk populations
- Dose reduction required in renal impairment
- Drug interactions: inhibits azathioprine and 6-mercaptopurine metabolism (these anticancer/immunosuppressant drugs are XOR substrates; allopurinol co-administration causes toxic accumulation)
- Starting dose should be low (100 mg/day or less) and titrated slowly to avoid precipitating an acute gout flare from rapid urate mobilization R
Febuxostat (Uloric):
A non-purine selective XOR inhibitor approved by the FDA in 2009.
Binds directly to the molybdopterin channel at the active site, blocking substrate access sterically and via hydrophobic interactions.
More potent than allopurinol at standard doses.
No dose reduction needed for mild-to-moderate renal impairment.
No HLA-B*5801 association.
Does not affect metabolism of azathioprine/6-mercaptopurine.
Important caveat: A 2018 FDA-required cardiovascular outcome trial (CARES) found a higher rate of cardiovascular death with febuxostat compared to allopurinol in patients with established cardiovascular disease.
The FDA subsequently added a black box warning; febuxostat is now indicated only for patients who cannot tolerate allopurinol.
The mechanism of the cardiovascular signal is debated; the data are complex and subsequent observational studies have not consistently confirmed it.
Topiroxostat:
Approved in Japan (not USA or Europe as of 2025).
A non-purine XOR inhibitor that forms a covalent linkage to the molybdenum via a hydroxylated 2-pyridine metabolite.
Hybrid competitive/covalent inhibition mechanism.
Uricosuric agents (not XOR inhibitors):
Probenecid, benzbromarone, and lesinurad inhibit URAT1, the renal urate reabsorption transporter.
They increase uric acid excretion rather than reducing production.
Useful when hyperuricemia is primarily excretion-driven (which is most patients).
Limited by reduced efficacy in renal impairment and risk of uric acid nephrolithiasis if used without adequate hydration.
Pegloticase:
Recombinant polyethylene glycol-conjugated uricase (from pig/baboon).
Converts uric acid to allantoin, bypassing the entire catabolic pathway.
Dramatically lowers uric acid and resolves tophi.
Reserved for refractory gout with tophaceous deposits not controlled by standard therapy.
Diet, Lifestyle, And Natural XO Inhibitors
Diet modifications that reduce XOR activity or uric acid load:
Reduce:
- Organ meats (liver, kidneys, heart): extremely high purine content
- Red meat and seafood (anchovy, sardines, herring, mackerel): high purine load
- Beer: purine-rich from yeast plus alcohol-driven excretion impairment
- Fructose and high fructose corn syrup: the ATP depletion mechanism is rapid and significant; avoid sugar-sweetened beverages especially
- Alcohol in general: ethanol is metabolized to acetaldehyde via alcohol dehydrogenase and also to acetyl-CoA, which competes with xanthine catabolism for NAD+; alcohol also reduces renal urate excretion
May help:
- Cherries and tart cherry juice: contain anthocyanins that inhibit XOR activity in vitro; multiple observational studies associate cherry consumption with reduced gout flare frequency; the mechanism likely involves both XOR inhibition and direct anti-inflammatory effects on NLRP3
- Vitamin C: uricosuric at doses above 500 mg/day; competes with urate for tubular reabsorption; meta-analyses show modest urate-lowering of approximately 0.35 mg/dL per 500 mg/day; effect is real but modest
- Dairy (low-fat): negatively associated with gout risk in epidemiological studies; casein and lactalbumin may have mild uricosuric effects
- Coffee: inversely associated with gout risk in multiple large cohorts; the mechanism is not primarily caffeine (decaffeinated coffee shows similar benefits); likely involves the chlorogenic acids and their metabolites as XOR inhibitors and uricosuric compounds
- Adequate hydration: critical for preventing renal urate crystal nucleation; target urine output of 2 L/day minimum for patients with nephrolithiasis history
Natural compounds with XOR inhibitory activity (research context, not clinical recommendations):
- Quercetin and other flavonoids: structurally similar to xanthine; competitive XOR inhibition in vitro; bioavailability and in vivo concentrations may limit clinical relevance
- Allopurinol-like xanthine analogues from various plant extracts (not commercially available as supplements)
- Luteolin, apigenin, kaempferol: in vitro XOR inhibition; clinical data limited
What To Stay Away From
- Starting allopurinol without HLA-B*5801 screening if you are of Han Chinese, Thai, or Korean ancestry: the risk of severe allopurinol hypersensitivity syndrome (Stevens-Johnson syndrome, toxic epidermal necrolysis) is dramatically elevated in individuals carrying this allele; cost of genetic testing is minimal compared to the potential catastrophic outcome; this screening is now part of standard care guidelines in Asian countries and is recommended by American College of Rheumatology guidelines for high-risk populations
- Starting XOR inhibitors at high doses acutely: rapid reduction of serum uric acid mobilizes crystal deposits, which can paradoxically trigger an acute gout flare; always start low (allopurinol at 100 mg/day or less, even 50 mg/day in CKD), titrate slowly over weeks to months to target, and use colchicine or NSAID prophylaxis for the first 3-6 months of urate-lowering therapy
- Combining allopurinol with azathioprine or 6-mercaptopurine without dose adjustment: these immunosuppressant drugs are XOR substrates; allopurinol inhibits their metabolism and causes toxic accumulation; the combination requires reducing azathioprine/6-mercaptopurine dose by approximately 75%; failure to do so can cause life-threatening myelosuppression
- Believing diet alone controls severe gout: dietary purines account for a minority of total uric acid production in most patients; dietary modification is useful and important but typically reduces serum urate by only 1-2 mg/dL maximum; patients with serum urate of 9-12 mg/dL cannot reach target on diet alone and require pharmacological urate-lowering therapy
- Ignoring uric acid in the context of metabolic syndrome: uric acid is not a gout-only biomarker; in the context of insulin resistance, hypertension, dyslipidemia, and central obesity, elevated uric acid is a marker of systemic metabolic dysfunction and XOR-driven oxidative stress; the cardiovascular and renal consequences of chronic hyperuricemia extend well beyond joint disease
- High fructose intake in the belief that "natural" fruit sugar is fine: the mechanism by which fructose elevates uric acid (fructokinase-driven ATP depletion) operates regardless of whether the fructose comes from high fructose corn syrup, table sugar, fruit juice, or dried fruit consumed in large quantities; whole fruit contains fiber that slows absorption and reduces the hepatic fructose load, making it much safer than fruit juice; the dose matters, and concentrated fructose sources are categorically different from eating whole fruit
Testing
Serum Uric Acid:
The primary diagnostic and monitoring test.
Normal range: 3.5-7.2 mg/dL in men, 2.6-6.0 mg/dL in women.
Gout treatment target: below 6.0 mg/dL; below 5.0 mg/dL in patients with tophi or frequent attacks.
Asymptomatic hyperuricemia above 8-9 mg/dL in men warrants consideration of urate-lowering therapy given cardiovascular and renal risk.
Serum uric acid fluctuates significantly; a single reading is insufficient; measure fasting, in the morning, on multiple occasions.
Important caveat: acute gout attacks can paradoxically lower serum uric acid transiently due to the inflammatory response; a normal level during an acute attack does not rule out gout.
24-Hour Urine Uric Acid:
Distinguishes overproducers from underexcretors.
Normal excretion: less than 800 mg/day on a regular diet.
Above 800 mg/day suggests overproduction; normal or low suggests underexcretion.
This distinction informs treatment choice: overproducers require XOR inhibition; underexcretors may respond to uricosuric agents.
Requires avoidance of purines in the 24-hour collection period for accurate baseline.
Comprehensive Metabolic Panel (CMP) and CBC:
Renal function (creatinine, BUN, eGFR): essential before starting allopurinol or febuxostat; both require monitoring in CKD; allopurinol dose must be reduced proportional to eGFR.
Liver enzymes: febuxostat is hepatically cleared; baseline and periodic monitoring.
CBC: if azathioprine is co-administered with allopurinol, regular CBC monitoring is mandatory for early detection of myelosuppression.
Fasting Insulin and HOMA-IR:
Serum XOR activity correlates with insulin resistance markers.
In a patient with hyperuricemia and metabolic syndrome features, quantifying insulin resistance is clinically useful both to understand the cause and to target the root driver (insulin resistance increases URAT1/GLUT9 expression, reducing renal urate excretion).
HLA-B*5801 Genetic Testing:
Before initiating allopurinol in patients of Han Chinese, Thai, or Korean ancestry.
A single-gene test that identifies the allele associated with severe hypersensitivity syndrome.
If positive, febuxostat or another alternative should be used.
Joint aspiration and synovial fluid analysis (clinical, not a lab order):
Gold standard for gout diagnosis.
Polarized light microscopy identifies needle-shaped, negatively birefringent MSU crystals.
This is how gout is definitively diagnosed and distinguished from calcium pyrophosphate deposition disease (pseudogout) and septic arthritis.
Necessary before initiating urate-lowering therapy in ambiguous cases.
Mechanisms Of Action
Simple:
- Xanthine is a purine base that sits at the crossroads of two important biological systems: it is the backbone that gets methylated by plants to make caffeine and chocolate compounds, and it is the molecule that the enzyme xanthine oxidase has to process before it can make uric acid; understanding both systems starts here.
- Every time your cells break down DNA, RNA, or ATP, they produce hypoxanthine and xanthine; the enzyme xanthine oxidase converts both of these to uric acid, and as a side effect it generates superoxide (a reactive oxygen species); normally this is manageable, but during a heart attack or stroke, xanthine builds up in the oxygen-starved tissue and then when blood flow returns, xanthine oxidase creates a sudden burst of superoxide that damages the recovering tissue.
- Caffeine, theophylline (in tea), and theobromine (in cocoa) all work primarily by blocking adenosine receptors; adenosine is a chemical signal your brain makes when it is tired, and methylxanthines block this signal without activating the receptor, which is why coffee makes you feel alert: you have chemically silenced the "you are tired" message without actually changing how tired you are.
- Gout happens when uric acid accumulates to the point where it crystallizes in joints; the crystals are recognized by immune cells as foreign invaders, which activate the NLRP3 inflammasome and trigger a massive inflammatory response that produces the intensely painful, swollen joint of a gout attack; xanthine oxidase is the enzyme that produces the uric acid, and XOR inhibitors like allopurinol work by blocking this final step.
- Fructose is uniquely dangerous for uric acid levels because its metabolism in the liver consumes ATP extremely rapidly; that depleted ATP is converted to AMP and then all the way down to uric acid; this is why high fructose corn syrup, table sugar, and fruit juice raise uric acid significantly faster than glucose does, and why the rise in gout over the 20th century tracked the rise in sugar consumption.
Advanced:
- The XDH-to-XO conversion as an ischemia timer: Under normoxic conditions, XOR exists predominantly as the XDH form, using NAD+ as electron acceptor and producing NADH rather than superoxide. During ischemia, two parallel processes arm a biochemical weapon: (1) ATP depletion drives purine catabolism through AMP → IMP → inosine → hypoxanthine, creating massive substrate accumulation; simultaneously, (2) ischemia-induced calcium influx, ROS from mitochondria, and protease activation convert XDH to XO via either reversible sulfhydryl oxidation (at mild hypoxia) or irreversible proteolytic cleavage (at severe hypoxia). The proteolytically converted XO cannot be reduced back to XDH; it is permanently committed to generating superoxide upon oxygen exposure. At reperfusion, the simultaneous availability of accumulated substrate (hypoxanthine/xanthine) and restored O2 drives the XO burst. The kinetics are important: XO's Km for oxygen is approximately 25 µM, well within physiological O2 tensions after reperfusion, meaning XO operates at near-maximum velocity immediately upon reoxygenation. Superoxide produced reacts with NO at rate constant k = 6.7 × 10^9 M^-1 s^-1, making peroxynitrite formation essentially diffusion-limited and certain wherever superoxide encounters the endothelial NO produced at reperfusion. R
- NLRP3 activation by XOR: two distinct pathways, one enzyme: XOR activates the NLRP3 inflammasome via two mechanistically distinct routes. First, XO-derived superoxide and H2O2 act as danger signals for NLRP3 assembly; ROS produced intracellularly by XO in macrophages activated by TLR ligands or crystal phagocytosis provide the mitochondrial ROS co-signal required for NLRP3 oligomerization. Second, when MSU crystals are present, they damage phagolysosomal membranes and release cathepsins, which provides the distinct danger signal triggering NLRP3 assembly; XO-derived ROS amplify this lysosomal damage pathway. The clinical evidence: febuxostat (but not allopurinol in all studies) prevents inflammasome assembly upstream of ASC speck formation and caspase-1 activation, reducing mature IL-1beta secretion; febuxostat specifically reduces intracellular superoxide, consistent with its tighter and more specific binding to XO blocking ROS generation more completely than allopurinol. XOR inhibitors also prevent NLRP3 activation in IBD mucosal tissue, where XO is upregulated in inflamed mucosa and drives IL-1beta and IL-18 production independently of urate crystals. R R
More Research
- Xanthine oxidoreductase exists in two interconvertible forms: XDH uses NAD+ as electron acceptor and produces NADH without generating ROS; XO uses O2 as electron acceptor and generates superoxide (O2-) and H2O2 as direct byproducts; conversion from XDH to XO occurs via reversible sulfhydryl oxidation or irreversible proteolytic cleavage, and this conversion is upregulated by hypoxia, inflammatory cytokines (IL-1, TNF-alpha, IFN-gamma), and ischemia, precisely the conditions where ROS generation is most harmful. R
- Methylxanthines (caffeine, theophylline, theobromine) are competitive nonselective adenosine receptor antagonists at all four receptor subtypes (A1, A2A, A2B, A3); caffeine and theophylline are roughly equipotent at adenosine receptors (Ki theophylline 6.7 µM vs caffeine 9.5 µM at A2A), but theophylline has more prominent bronchodilator and PDE inhibitory effects relative to CNS stimulant effects; theobromine is a weak adenosine receptor antagonist with primarily cardiovascular and bronchopulmonary activity. R
- Fructose raises uric acid via an ATP depletion mechanism that does not apply to glucose: fructokinase phosphorylates fructose to fructose-1-phosphate without product inhibition, rapidly consuming hepatic ATP; the resulting AMP accumulation drives purine catabolism through IMP → inosine → hypoxanthine → xanthine → uric acid; fructose also upregulates XOR transcription and de novo purine synthesis, creating a triple hit on uric acid production; high-fructose diets in animal models induce hyperuricemia, hypertension, insulin resistance, and hypertriglyceridemia, all of which are significantly blunted by allopurinol. R
- Humans and higher primates lost functional uricase approximately 15-20 million years ago through two independent mutational events; this renders uric acid the end product of purine catabolism rather than allantoin; as a result, normal human serum uric acid levels (3-7 mg/dL) are roughly 5-10 times higher than in other mammals with intact uricase; this evolutionary change may have provided antioxidant benefits but created unique vulnerability to hyperuricemia, gout, and urate-driven inflammatory disease. R
- XOR inhibition with allopurinol in patients with dilated cardiomyopathy given intracoronarily reduced myocardial oxygen consumption and improved left ventricular function, while probenecid (a uricosuric that lowers uric acid without inhibiting XOR) did not produce the same effect; this dissociation demonstrates that the cardiac benefit of allopurinol in heart failure is attributable to XOR-derived ROS suppression, not urate reduction per se, and supports XOR as an independent therapeutic target in cardiac oxidative stress. R
- XO is upregulated in the inflamed intestinal mucosa of both UC and Crohn's disease patients; XO overexpression correlates with increased NLRP3 inflammasome activation, caspase-1 activity, and IL-1beta and IL-18 production; ex vivo treatment of inflamed biopsies with allopurinol or febuxostat reduces NLRP3 assembly, caspase-1 activity, and both cytokines, suggesting XOR inhibition as a potential adjunct anti-inflammatory strategy in IBD independent of gout. R
- Estrogen suppresses hepatic XOR activity by approximately 40% compared to males, and ovariectomy restores activity to male levels; estrogen also suppresses the renal urate reabsorption transporters URAT1 and GLUT9 while upregulating the intestinal efflux transporter ABCG2; these effects collectively explain why premenopausal women have substantially lower serum uric acid than men, why gout is rare in premenopausal women, and why gout incidence in women rises sharply after menopause to approach male incidence rates.
- Allopurinol hypersensitivity syndrome (AHS), a potentially fatal reaction including Stevens-Johnson syndrome and toxic epidermal necrolysis, is strongly associated with the HLA-B*5801 allele; the allele frequency in Han Chinese and Thai populations is 6-8% compared to less than 0.1% in European populations; genetic screening before allopurinol initiation is now standard of care in high-risk populations and is included in American College of Rheumatology and European League Against Rheumatism gout guidelines. R
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Electrolyte Complex
1 scoop/day
CoQ10
200mg/day
Magnesium Glycinate
400mg at bedtime