NAD+, NADH, NADP+, And NADPH: The Redox Molecules Running Every Cell You Have
By Jacob Gordon, INHC, FMT-CThis article contains affiliate links. As an Amazon Associate, MyBioHack earns from qualifying purchases at no extra cost to you. We only link products we research and stand behind.
These four molecules are the reason your cells can make energy, repair DNA, neutralize free radicals, synthesize fat, and stay alive.
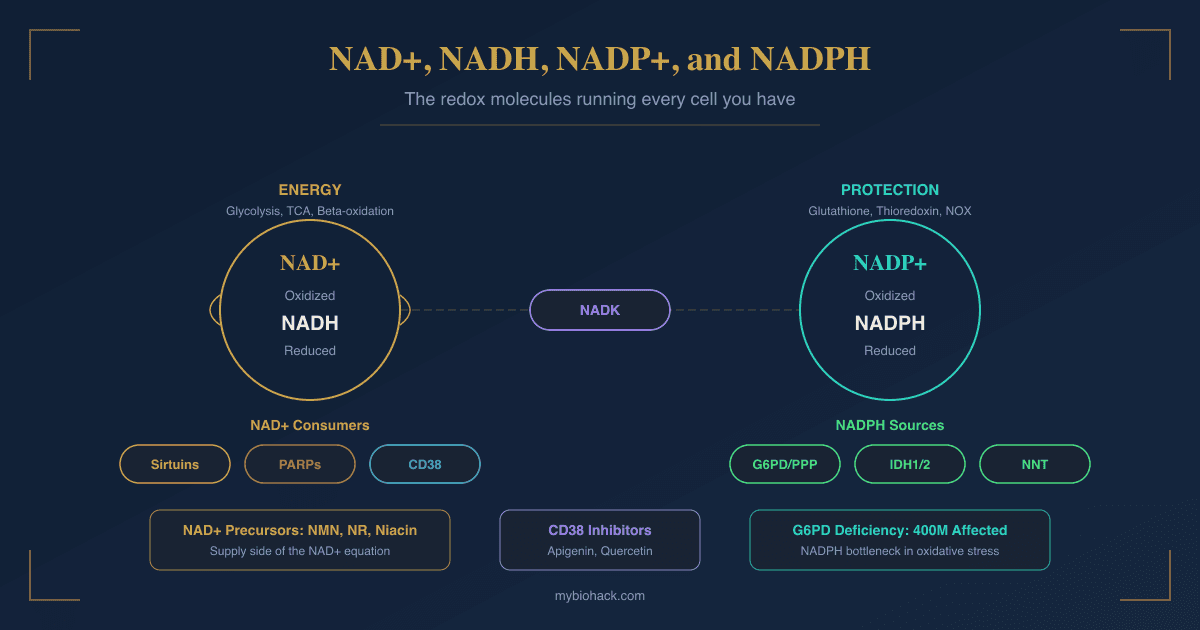
NAD+ and NADH are the oxidized and reduced forms of nicotinamide adenine dinucleotide.
NADP+ and NADPH are the phosphorylated versions of that same molecule, separated from NAD+/NADH by a single phosphate group.
That single phosphate group changes everything about what the molecule does.
NAD+/NADH operates primarily in catabolic metabolism: accepting electrons from fuel oxidation and feeding them into the electron transport chain to make ATP.
NADP+/NADPH operates primarily in anabolic and antioxidant metabolism: providing the reducing power to build things, detoxify ROS, and protect cells from oxidative damage.
Understanding both pairs, their separate functions, where they are made, what consumes them, and how they decline with age and disease, is foundational to understanding why so many chronic conditions share a common biochemical thread.

What NAD+ Actually Is: Structure And Chemistry
Nicotinamide adenine dinucleotide (NAD+) is a dinucleotide coenzyme composed of two mononucleotides joined by a pyrophosphate bridge: adenosine monophosphate (AMP) on one side and nicotinamide mononucleotide (NMN) on the other.
The nicotinamide ring in NMN carries the positively charged nitrogen that gives NAD+ its oxidized form.
When NAD+ accepts a hydride ion (H-, which is a proton plus two electrons), that positively charged nitrogen is neutralized, and the molecule becomes NADH: its fully reduced form.
This hydride transfer is the core chemical event that underlies most of energy metabolism.
The reaction is reversible, which is what makes the coenzyme useful: NAD+ acts as an electron acceptor, NADH acts as an electron donor, and the cell cycles between them continuously.
NADP+ is structurally identical to NAD+ with one exception: a phosphate ester is attached to the 2'-hydroxyl group of the adenosine ribose.
This structural change creates a separate coenzyme pool that is functionally segregated from NAD+/NADH.
In practical terms:
- NAD+/NADH ratio reflects catabolic state; high NAD+/NADH means the cell is actively oxidizing fuel
- NADP+/NADPH ratio reflects redox and anabolic state; high NADPH means the cell has reducing power available for biosynthesis and antioxidant defense
These ratios are regulated independently in the cytoplasm and mitochondria.
The mitochondrial and cytoplasmic NAD+ pools do not freely exchange because the inner mitochondrial membrane is impermeable to NAD+; each compartment maintains its own pool, replenished and consumed by compartment-specific enzymes. R
The NAD+/NADH Redox Couple: Energy Metabolism
Every major fuel oxidation pathway in the cell generates NADH by donating electrons to NAD+.
Glycolysis (cytoplasm): Glyceraldehyde-3-phosphate dehydrogenase oxidizes G3P to 1,3-bisphosphoglycerate, reducing one NAD+ to NADH per reaction (two per glucose molecule). R
Pyruvate dehydrogenase (mitochondrial matrix): Pyruvate is converted to acetyl-CoA with release of CO2, generating one NADH.
TCA cycle (Krebs cycle, mitochondrial matrix): Three of the eight steps in the TCA cycle generate NADH (isocitrate dehydrogenase, alpha-ketoglutarate dehydrogenase, malate dehydrogenase).
One step generates FADH2 (succinate dehydrogenase, which is also Complex II of the electron transport chain).
Beta-oxidation (mitochondrial matrix): Each cycle of fatty acid beta-oxidation generates one NADH and one FADH2 per acetyl-CoA cleaved.
The electron transport chain: NADH donates its electrons to Complex I (NADH dehydrogenase), the first protein complex of the inner mitochondrial membrane.
The electron transfer drives proton pumping across the inner membrane, building the proton gradient that ATP synthase (Complex V) uses to phosphorylate ADP to ATP.
Each NADH yields approximately 2.5 ATP.
FADH2 donates its electrons to Complex II, bypassing Complex I, and yields approximately 1.5 ATP per molecule.
The critical constraint: When NADH accumulates faster than it can be oxidized by Complex I (which happens under high energy availability, poor mitochondrial function, or in hypoxia), NAD+ becomes rate-limiting.
Glycolysis stalls because the glyceraldehyde-3-phosphate dehydrogenase step requires NAD+ and cannot proceed with only NADH available.
This is why lactate fermentation exists: lactate dehydrogenase (LDH) regenerates NAD+ from NADH by converting pyruvate to lactate, allowing glycolysis to continue even without oxidative phosphorylation.
This is exactly what happens in rapidly dividing cancer cells (the Warburg effect) and in hypoxic tissue.
Beyond Redox: NAD+ As A Consumed Substrate
NAD+ is unique among coenzymes in that it is not merely a recycled carrier of electrons.
Three major classes of enzymes cleave NAD+ as a substrate, consuming it stoichiometrically: the molecule is destroyed, and nicotinamide (NAM) is released as a byproduct. R
This is why NAD+ must be continuously resynthesized.
In the redox reactions of glycolysis and the TCA cycle, NAD+ is simply reduced to NADH and then re-oxidized back to NAD+ by the electron transport chain.
No net NAD+ is consumed.
But when a sirtuin deacetylates a protein, when PARP1 responds to a DNA break, or when CD38 generates calcium-signaling metabolites, that NAD+ molecule is gone.
This distinction matters enormously for understanding why NAD+ declines with aging, inflammation, and oxidative stress: the non-redox consumers are upregulated by all of those conditions and they permanently consume NAD+.
Sirtuins
Sirtuins (SIRT1-7) are a family of NAD+-dependent deacylases that remove acetyl (and other acyl) groups from lysine residues on target proteins, transferring the acyl group to the ADP-ribose fragment of NAD+ and releasing NAM and O-acetyl-ADP-ribose. R
They are sometimes called "longevity enzymes" because the yeast Sir2 ortholog extends lifespan when overexpressed, and because sirtuin activity promotes many processes associated with healthy aging: mitochondrial biogenesis, antioxidant defense, DNA repair, inflammation resolution, and metabolic flexibility.
The seven mammalian sirtuins have distinct subcellular localizations and substrates:
- SIRT1: Nucleus/cytoplasm; deacetylates histones H3K9, H3K14, H4K16, and transcription factors including p53, NF-kB, FOXO proteins, and PGC-1alpha; master regulator of metabolism, circadian rhythm, and stress response
- SIRT2: Cytoplasm; deacetylates alpha-tubulin; involved in cell cycle, mitosis, and neurodegeneration
- SIRT3: Mitochondria; deacetylates metabolic enzymes; regulates ATP production, ROS detoxification, and fatty acid oxidation
- SIRT4: Mitochondria; primarily a lipoamidase and ADP-ribosyltransferase; regulates fatty acid oxidation and insulin secretion
- SIRT5: Mitochondria; desuccinylase, demalonylase, deglutarylase; regulates the TCA cycle and urea cycle
- SIRT6: Nucleus; deacetylates H3K9ac and H3K56ac; maintains genomic stability, telomere integrity, and DNA double-strand break repair
- SIRT7: Nucleolus; regulates rRNA transcription, ribosome biogenesis, and chromatin organization R
Key SIRT1 axis: SIRT1 deacetylates PGC-1alpha, activating it to drive mitochondrial biogenesis.
SIRT1 also deacetylates LKB1, activating AMPK.
SIRT1 suppresses NF-kB inflammatory signaling by deacetylating the p65 subunit.
SIRT1 deacetylates p53, reducing apoptosis.
These effects collectively explain why raising NAD+ (and thus increasing SIRT1 activity) has anti-inflammatory, pro-mitochondrial, and pro-survival effects across multiple tissue types.
The NAD+-sirtuin-NAMPT axis and circadian rhythm: NAMPT (nicotinamide phosphoribosyltransferase), the rate-limiting enzyme of NAD+ salvage synthesis, is itself a SIRT1 target and exhibits robust circadian oscillation.
SIRT1 deacetylates and activates CLOCK/BMAL1, the core circadian transcription factors.
This creates a feedback loop: NAD+ drives SIRT1 which drives circadian machinery which drives NAMPT which regenerates NAD+.
Disruption of this loop by chronic inflammation, sleep disruption, or aging accelerates NAD+ decline. R
PARPs And DNA Repair
Poly(ADP-ribose) polymerases (PARPs) are a family of 17 enzymes that transfer ADP-ribose units from NAD+ onto target proteins, forming poly-ADP-ribose (PAR) chains.
PARP1 is the most NAD+-consuming and most studied family member.
When PARP1 detects a DNA single-strand break, it binds the break site and begins PARylating itself and nearby histones, signaling for the recruitment of DNA repair machinery. R
Under moderate DNA damage, PARP1 activation is essential for genome integrity.
Under severe DNA damage (from radiation, alkylating agents, oxidative stress, or viral infection), PARP1 becomes hyperactivated and can consume so much NAD+ so rapidly that cellular NAD+ pools are depleted within minutes.
This triggers a catastrophic metabolic collapse: ATP synthesis fails because the electron transport chain cannot proceed without sufficient NADH regeneration, and cells die through a necrotic pathway called parthanatos (PARP-dependent cell death).
Paradoxically, PARP1 and SIRT1 compete directly for NAD+.
High PARP1 activity depletes the NAD+ that SIRT1 needs to operate, resulting in reduced sirtuin activity and the accumulation of acetylated (inactivated) sirtuin targets.
This PARP-sirtuin competition is a core mechanism by which oxidative stress and DNA damage suppress mitochondrial function and accelerate aging:
PARP1 is activated by ROS-induced DNA damage, PARP1 consumes NAD+, SIRT1 loses NAD+ substrate, PGC-1alpha remains acetylated/inactive, mitochondrial biogenesis is suppressed, cells become less able to clear ROS, more DNA damage occurs, more PARP1 activation follows. R
PARP2 directly inhibits SIRT1 transcription, adding a second layer of suppression.
PARP inhibitors (olaparib, niraparib, rucaparib) are approved cancer drugs that exploit this pathway.
In BRCA1/2-deficient tumors, PARP inhibition is synthetically lethal because the cancer cells cannot repair double-strand breaks through homologous recombination and are now also blocked from repairing single-strand breaks.
CD38: The NAD+ Drainer
CD38 is an ectoenzyme and NAD+ glycohydrolase that is a major consumer of NAD+ in mammalian tissues.
It is expressed on immune cells (particularly T cells, B cells, NK cells, and macrophages), on the outer plasma membrane surface (as an ectoenzyme), and intracellularly.
CD38 converts NAD+ to cyclic ADP-ribose (cADPR), which triggers calcium channel opening and intracellular Ca2+ release, and to ADPR and other metabolites. R
This makes CD38 a master regulator of calcium signaling in immune cells.
The problem is that CD38 has a lower Km for NAD+ than most other NAD+-consuming enzymes, meaning it has high affinity for NAD+ even at low concentrations.
CD38 is therefore a disproportionate consumer that can deplete NAD+ even when overall NAD+ levels are relatively low.
CD38 expression increases dramatically with aging and inflammation.
This is now recognized as a primary mechanism of the NAD+ decline seen in aging: chronically elevated inflammation (inflammaging) drives CD38 upregulation in senescent cells and immune cells, which then depletes NAD+ in neighboring cells via the connexin 43 hemichannels that supply CD38 with extracellular NAD+.
PARP1 knockout and CD38 knockout mice both show elevated NAD+ levels and improved SIRT1 activity, confirming that these two consumers are the primary brakes on NAD+ availability in vivo. R
Apigenin (a flavonoid found in parsley, chamomile, and celery) is a documented CD38 inhibitor with reasonable human bioavailability.
Quercetin also inhibits CD38, though less potently than apigenin.
This is one of the most mechanistically coherent cases for using these flavonoids to support NAD+ biology.
SARM1: Axonal Degeneration
SARM1 (sterile alpha and TIR motif-containing protein 1) is a NAD+ glycohydrolase expressed specifically in neurons.
Under normal conditions, SARM1 is held in an autoinhibited state.
Following axon injury, metabolic stress, or pathogen-associated signals, SARM1 is activated and rapidly consumes NAD+ in the axon, triggering Wallerian degeneration: the programmatic self-destruction of the distal axon following injury. R
SARM1 is relevant to traumatic brain injury, peripheral neuropathy, chemotherapy-induced neuropathy, and neurodegenerative diseases where axonal loss is a feature.
SARM1 inhibitors are in early-stage drug development.
NADP+/NADPH: The Anabolic And Antioxidant Pool
NAD+ kinase (NADK) is the sole enzyme responsible for creating NADP+ from NAD+.
NADK transfers a phosphate group from ATP to the 2'-hydroxyl of the adenosine ribose of NAD+, producing NADP+. R
NADK is expressed in both cytoplasm (NADK1) and mitochondria (MNADK).
NADK activity is therefore the critical branch point that determines how much of the NAD+ pool is directed toward the NADP+/NADPH system.
Crucially, NADK converts NAD+ to NADP+ but cannot convert NADH to NADPH.
The mitochondrial transhydrogenase enzyme performs that conversion in mitochondria specifically (NADH + NADP+ to NAD+ + NADPH), driven by the proton gradient.
This transhydrogenase is the primary source of mitochondrial NADPH.
Cellular NADP+/NADPH ratio vs. NAD+/NADH ratio:
The NADP+/NADPH ratio is kept very low in healthy cells, meaning most of the NADP pool is maintained in the reduced NADPH form.
This is opposite to the NAD+/NADH ratio, which is kept relatively high (more oxidized NAD+).
This makes functional sense: NADPH needs to be available continuously for antioxidant reactions that can be triggered at any moment, while NAD+ needs to be available to accept electrons from fuel oxidation.
Where NADPH Comes From: G6PD And The Pentose Phosphate Pathway
Glucose-6-phosphate dehydrogenase (G6PD) is the rate-limiting enzyme of the pentose phosphate pathway (PPP) and the primary cytoplasmic source of NADPH.
G6PD catalyzes the first reaction of the PPP oxidative phase: conversion of glucose-6-phosphate to 6-phosphogluconolactone, simultaneously reducing NADP+ to NADPH. R
The second enzyme in the oxidative branch, 6-phosphogluconate dehydrogenase (6PGD), generates a second NADPH per glucose-6-phosphate committed to the pathway.
The non-oxidative phase then interconverts the remaining carbon skeletons into ribose-5-phosphate (needed for nucleotide synthesis) and feeds fructose-6-phosphate and glyceraldehyde-3-phosphate back into glycolysis.
Under oxidative stress, cells rapidly upregulate PPP flux and G6PD activity: glucose is diverted from glycolysis toward the PPP to regenerate NADPH for antioxidant enzymes.
G6PD activity is regulated both transcriptionally (by NRF2, JAK-STAT, mTOR, Wnt) and post-translationally (by phosphorylation and acetylation in response to stress signals).
Acute increases in G6PD activity can involve physical translocation of the enzyme to the plasma membrane, allowing entering glucose to be immediately shunted into the PPP. R
G6PD deficiency is the most common human enzyme deficiency worldwide, affecting approximately 400 million people.
It is X-linked and provides partial protection against malaria because G6PD-deficient red blood cells provide a hostile environment for Plasmodium falciparum, which depends heavily on host NADPH for its own growth.
Clinical consequences of G6PD deficiency:
- Hemolytic anemia triggered by oxidative stress (from infections, fava beans, antimalarials like primaquine, dapsone, high-dose vitamin C)
- Impaired neutrophil oxidative burst (NADPH oxidase requires NADPH to generate the superoxide used to kill pathogens)
- Neonatal jaundice
Additional NADPH Sources
G6PD is the dominant cytoplasmic source of NADPH, but several other enzymes contribute: R
Isocitrate dehydrogenase 1 (IDH1): Cytoplasmic; converts isocitrate to alpha-ketoglutarate using NADP+, generating NADPH; serves as a backup NADPH source when G6PD is impaired.
Malic enzyme 1 (ME1): Cytoplasmic; converts malate to pyruvate, generating NADPH; links NADPH production to mitochondrial metabolite shuttling.
Isocitrate dehydrogenase 2 (IDH2): Mitochondrial; major source of mitochondrial NADPH.
Malic enzyme 3 (ME3): Mitochondrial.
Glutamate dehydrogenase (GLUD1): Mitochondrial; produces NADPH while converting glutamate to alpha-ketoglutarate.
Mitochondrial transhydrogenase (NNT): Mitochondrial inner membrane; the primary mitochondrial NADPH source; transfers reducing equivalents from NADH to NADPH driven by the proton gradient.
Methylenetetrahydrofolate dehydrogenase 1 (MTHFD1/MTHFD2): Linked to the folate cycle; relevant specifically for MTHFR variants and one-carbon metabolism.
The relative importance of each source is tissue-specific and context-dependent.
IDH1 is a key backup in cells where G6PD is limiting.
IDH1 mutations that convert this NADPH-generating reaction to an NADPH-consuming one (producing the oncometabolite 2-hydroxyglutarate instead of alpha-ketoglutarate) are found in gliomas, AML, and chondrosarcomas, and they simultaneously deplete cellular NADPH and generate an epigenetic disruptor.
What NADPH Actually Does
1. Glutathione system (primary antioxidant network): Glutathione peroxidase (GPx) uses reduced glutathione (GSH) to neutralize hydrogen peroxide (H2O2) and lipid peroxides, converting GSH to oxidized glutathione (GSSG).
Glutathione reductase then regenerates GSH from GSSG, using NADPH as the electron donor.
Without NADPH, glutathione becomes trapped in its oxidized (GSSG) form, antioxidant capacity collapses, and ROS accumulate. R
This is why G6PD-deficient red blood cells cannot withstand oxidative stress: they cannot regenerate GSH from GSSG because they cannot generate NADPH without G6PD.
2. Thioredoxin system (parallel antioxidant network): Thioredoxin (Trx) is a small protein that directly reduces protein disulfides and peroxides.
Thioredoxin reductase (TrxR) regenerates oxidized thioredoxin using NADPH.
The thioredoxin system runs in parallel to the glutathione system and is particularly important in mitochondria. R
3. Catalase stabilization: Catalase directly converts H2O2 to water and oxygen.
NADPH binds to catalase and prevents its inactivation by high H2O2 concentrations (compound I formation).
NADPH does not donate electrons in this reaction; it stabilizes the enzyme's active conformation.
4. NADPH oxidases (NOX enzymes): controlled ROS production: This is the counterintuitive function: NADPH is also the substrate for NADPH oxidases (NOX1-5, DUOX1-2), which deliberately generate superoxide as a signaling molecule and pathogen-killing agent.
In neutrophils and macrophages, the respiratory burst that kills engulfed pathogens is driven by NOX2 using NADPH to generate massive amounts of superoxide in the phagolysosome. R
In signaling contexts across all cell types, low-level NOX-derived ROS function as second messengers activating NF-kB, MAPK, and NRF2 pathways.
The dual role of NADPH in both generating ROS (via NOX) and neutralizing ROS (via glutathione and thioredoxin systems) means NADPH availability sits at the center of redox balance.
This has practical implications: simply increasing NADPH without understanding which enzymes are most active in a particular context could theoretically increase both oxidant production and antioxidant capacity.
5. Fatty acid synthesis: Every round of fatty acid chain elongation by fatty acid synthase (FAS) requires two NADPH molecules (one at the ketoacyl reductase step, one at the enoyl reductase step).
Synthesis of palmitate (16 carbons) from acetyl-CoA requires 14 NADPH molecules.
Cholesterol synthesis (via HMG-CoA reductase and downstream steps) also consumes NADPH.
This is why rapidly proliferating cells upregulate the PPP: they need NADPH not just for antioxidant defense but for the membrane lipid synthesis required for cell division. R
6. Cytochrome P450 enzymes: The hepatic CYP450 drug-metabolizing enzyme superfamily uses NADPH to drive their reductive-oxidative cycles.
Phase I biotransformation of drugs, steroids, vitamin D, and xenobiotics depends on NADPH.
Nitric oxide synthase (iNOS, eNOS, nNOS) also requires NADPH to convert arginine to NO.
7. DNA synthesis: Ribonucleotide reductase converts ribonucleotide diphosphates to deoxyribonucleotide diphosphates, fueling DNA replication, using NADPH as the electron donor.
Dihydrofolate reductase (DHFR) also requires NADPH, linking NADPH to folate metabolism and methylation. R
How NAD+ Is Made: The Three Biosynthesis Pathways
NAD+ is synthesized through three distinct routes, which differ in their starting material and enzymatic steps.
1. De novo synthesis: the kynurenine pathway
Starting material: dietary tryptophan (Trp).
Tryptophan is converted through the kynurenine pathway to quinolinic acid (QUIN), then to nicotinic acid mononucleotide (NAMN), and eventually to NAD+.
This pathway requires multiple enzymatic steps including IDO1 (indoleamine 2,3-dioxygenase), which is induced by inflammation.
De novo synthesis from tryptophan requires approximately 60mg tryptophan to produce 1mg niacin equivalent, making it an inefficient pathway under most conditions.
It is, however, relevant in the context of chronic inflammation: IDO1 upregulation diverts tryptophan toward kynurenine pathway intermediates, some of which are neurotoxic (quinolinic acid activates NMDA receptors), at the expense of serotonin synthesis. R
2. Preiss-Handler pathway
Starting material: nicotinic acid (NA), the form of niacin without an amide group (niacin/vitamin B3 in the older sense).
NA is converted to NAMN, then to NAAD, then to NAD+.
The final step, NAAD to NAD+, is catalyzed by NAD synthetase, which requires glutamine as nitrogen donor.
This pathway is used by dietary niacin obtained primarily from animal foods and fortified grain products.
Nicotinic acid at pharmacological doses (1500-3000 mg/day) produces the flushing reaction through GPR109A activation on skin Langerhans cells, prostaglandin D2 release, and vasodilation.
Extended-release niacin reduces this flushing but does not eliminate it.
3. Salvage pathway (the dominant pathway in most cells)
This is the primary means by which mammalian cells maintain their NAD+ pools.
It recycles nicotinamide (NAM), the byproduct of every sirtuin, PARP, and CD38 reaction.
The rate-limiting enzyme is NAMPT (nicotinamide phosphoribosyltransferase), which converts NAM to NMN using PRPP (5-phosphoribosyl-1-pyrophosphate) as the phosphoribosyl donor.
NMNAT1/2/3 then converts NMN to NAD+ (in nucleus, cytoplasm, and mitochondria respectively).
NMN and NR (nicotinamide riboside) from dietary sources also enter through the salvage pathway:
- NR enters cells via equilibrative nucleoside transporters and is phosphorylated to NMN by NR kinase (NRK1/2)
- NMN enters cells via the SLC12A8 transporter and is directly converted to NAD+ by NMNAT
The salvage pathway is essential for life: NAMPT knockout mice die before birth.
NAMPT activity is circadian-regulated and is inhibited by high NAM concentrations (product inhibition), providing built-in feedback control. R
NNMT (nicotinamide N-methyltransferase) is an important shunt in this pathway: it methylates NAM to form methyl-NAM, consuming NAM and diverting it away from the salvage pathway.
NNMT upregulation (driven by high-fat diet or chronic inflammation) therefore reduces NAD+ synthesis by reducing available NAM for NAMPT.
It also consumes SAM-e (S-adenosylmethionine) methyl groups, potentially creating tension with methylation-dependent processes.
NAD+ Decline In Aging And Disease
NAD+ levels in humans decline steadily with age across multiple tissues, including blood, skin, muscle, liver, and brain. R
The decline is not uniform: it is estimated at 30-50% between middle and old age in some tissues, with brain and muscle showing particularly steep declines.
The mechanisms of decline are now reasonably well understood and they interact:
CD38 upregulation: Senescent cells and activated macrophages secrete inflammatory cytokines that upregulate CD38 on surrounding cells.
CD38 then consumes NAD+ at rates that outpace NAMPT-mediated regeneration.
PARP1 hyperactivation: Accumulated DNA damage with age (from ROS, mitochondrial DNA errors, replication stress) chronically activates PARP1, depleting NAD+ faster than it can be regenerated.
NAMPT downregulation: Chronic inflammation and circadian disruption reduce NAMPT expression and activity, slowing the salvage pathway.
NNMT upregulation: Methylation of NAM diverts it away from recycling.
Reduced tryptophan availability: IDO1 upregulation in chronic inflammation and immune activation diverts tryptophan toward kynurenine rather than serotonin, and downstream QUIN accumulation is neurotoxic at elevated levels.
Consequences of NAD+ decline:
The downstream consequences map closely onto the hallmarks of aging:
- Reduced sirtuin activity: impaired mitochondrial biogenesis, accumulation of acetylated metabolic enzymes, loss of circadian robustness, impaired DNA repair (SIRT6 depletion accelerates aging markedly)
- Reduced PARP efficiency: more DNA damage accumulation despite PARP being activated, because NAD+ depletion limits how long PARP can remain active
- Reduced mitochondrial function: NADH cannot be efficiently oxidized, Complex I slows, ROS generation from the electron transport chain increases
- Impaired immune function: T cell activation and NK cell killing both require NAD+ for CD38-mediated calcium signaling
- Sarcopenia: NAD+ is required for muscle satellite cell activation and differentiation; NAD+ decline contributes to impaired muscle regeneration
- Cognitive decline: neurons are particularly dependent on NAD+ for energy metabolism and DNA repair; SIRT1 and SIRT6 are required for synaptic plasticity R
NAD+ Precursor Supplementation: NMN, NR, Niacin
The rationale for NAD+ precursor supplementation is straightforward: if NAD+ levels decline with age and this decline causes dysfunction, then restoring NAD+ should improve function.
The animal data supporting this is compelling.
NMN supplementation in aged mice mitigates multiple age-associated physiological declines including muscle function, bone density, eye function, energy metabolism, and immune function.
NR supplementation in aged mice improves mitochondrial biogenesis and function.
The human clinical trial data is more mixed, and an honest account of it matters.
Consistent Finding: NAD+ Levels Rise
Multiple human RCTs confirm that oral NMN and NR supplementation reliably raise blood NAD+ levels.
NMN (250-500 mg/day) raises whole blood NAD+ levels approximately 2-6 fold depending on baseline.
NR (1000-2000 mg/day) raises blood NAD+ levels 1.4-2 fold.
Acute NR supplementation (900 mg single dose) increased brain NAD+ concentrations in humans measured by 7 Tesla MRI spectroscopy. R
Human Clinical Trial Outcomes: Honest Summary
The 2024 meta-analysis of NMN supplementation trials found that NMN significantly elevated blood NAD+ levels but most clinically relevant metabolic outcomes (fasting glucose, triglycerides, total cholesterol, LDL, HDL) were not significantly different from placebo across 12 RCTs with 513 participants. R
The heterogeneity across studies is partly explained by large individual variation in baseline NAD+ levels.
NMN-treated subjects who had low baseline NAD+ showed more robust functional improvements than those with already adequate levels.
Positive signals in specific populations:
NMN 250 mg/day for 12 weeks in healthy older adults (65-75 years) produced faster 4-meter walking time, improved sleep quality, and higher NAD+ metabolites compared to placebo. R
NMN improved aerobic capacity and oxygen utilization in middle-aged runners in a Japanese RCT.
NR (2000 mg/day) for 24 weeks in long-COVID patients with persistent cognitive symptoms produced improvements in cognition and fatigue scores alongside significant NAD+ elevation. R
NMN reduced arterial stiffness, diastolic blood pressure, LDL, and body weight in healthy middle-aged adults over 12 weeks.
Comparing NMN vs. NR
Both enter the salvage pathway.
NR requires phosphorylation by NRK to become NMN, then NMNAT converts NMN to NAD+.
NMN enters cells directly via the SLC12A8 transporter (bypassing the NRK step) and is converted to NAD+ by NMNAT.
Whether NMN's slightly more direct route provides a meaningful pharmacokinetic advantage in humans is debated.
NMN is generally more expensive than NR.
Both are safe at typical supplementation doses (250-1000 mg/day for NMN, 300-2000 mg/day for NR) based on current trial data.
Nicotinamide (NAM) vs. Nicotinic Acid (NA)
Plain nicotinamide is inexpensive and reliably raises NAD+, but at doses above 500 mg/day chronically it can inhibit sirtuins by product-inhibition feedback (since NAM is the sirtuin reaction product).
This makes high-dose nicotinamide an imperfect NAD+ precursor: it raises total NAD+ but simultaneously inhibits the very enzymes you want to activate.
Nicotinic acid (niacin) causes flushing at therapeutic doses, which limits tolerance.
Neither is a preferred option compared to NMN or NR for sirtuin activation, though they are cheaper for general NAD+ elevation without sirtuin-specific goals.
CD38 Inhibition As A Complementary Strategy
Because CD38 upregulation with aging is a primary driver of NAD+ decline, inhibiting CD38 while also supplying precursors is a more complete strategy than precursors alone.
Apigenin: the most potent natural CD38 inhibitor identified; found in parsley, chamomile tea, and celery.
Apigenin: 50-100mg daily.
Quercetin: also inhibits CD38 and has additional effects on NAMPT activation.
Quercetin: 500-1000mg daily; note quercetin inhibits CYP3A4 at higher doses.
Lifestyle Inputs That Raise NAD+
These are not supplements but they reliably affect NAD+ biology:
Exercise: Acute aerobic exercise increases NAD+ turnover and SIRT1 activity.
Chronic aerobic training upregulates NAMPT expression in muscle and liver.
Combined aerobic exercise and NMN supplementation shows additive increases in NAD+ compared to either alone.
Caloric restriction and fasting: Fasting shifts the NAD+/NADH ratio toward higher NAD+ by reducing NADH production from substrate oxidation (less fuel available) while NAMPT-driven synthesis continues.
SIRT1 is activated under CR partly through this NAD+ increase and partly through reduced inhibitory acetylation.
Reducing inflammation: Since chronic inflammation upregulates CD38 and PARP1, any intervention that reduces inflammatory burden (treating infections, improving gut barrier, reducing visceral fat, resolving autoimmune drivers) will indirectly support NAD+ levels.
Sleep: Circadian disruption degrades CLOCK/BMAL1-mediated NAMPT oscillation; consistent sleep timing supports the circadian NAD+ synthesis cycle.
What To Stay Away From
- High-dose nicotinamide (NAM) above 500mg/day chronically as a sirtuin-activation strategy: it raises total NAD+ but NAM is the sirtuin inhibitor in the feedback loop; it may be appropriate for general NAD+ repletion but is counterproductive if the goal is to maximize SIRT1 or other sirtuin activity
- Alcohol: ethanol metabolism generates large amounts of NADH via alcohol dehydrogenase and aldehyde dehydrogenase, driving up the NADH/NAD+ ratio; this inhibits gluconeogenesis (hypoglycemia), activates lipogenesis (fatty liver), and consumes NAD+ that should be available for sirtuins and PARPs
- Chronic high-dose niacin above 3000mg/day without medical supervision: at very high doses, niacin causes glucose intolerance, hepatotoxicity, and rare cases of rhabdomyolysis; clinical pharmacological use for dyslipidemia has largely been displaced by statins
- Assuming NMN/NR supplements will replicate caloric restriction or exercise: they reliably raise NAD+, but most human trials show modest functional effects at currently tested doses; they are not substitutes for the lifestyle inputs that drive NAD+ biology most powerfully; the animal evidence that drives commercial excitement involved much larger relative dose equivalents than typical human supplementation
- G6PD-deficient individuals taking oxidizing supplements without awareness of their deficiency: high-dose vitamin C, alpha-lipoic acid at very high doses, methylene blue, and some antimalarials can precipitate hemolytic crises in G6PD-deficient individuals by generating oxidative stress that depleted NADPH cannot buffer; G6PD deficiency should be ruled out before aggressive oxidative or antioxidant supplementation in populations where it is prevalent (sub-Saharan African, Mediterranean, Middle Eastern, and East Asian ancestries)
Testing
There is currently no widely available clinical test that measures tissue NAD+ levels directly.
Blood NAD+ measurement is available through specialty labs but has significant limitations: whole blood NAD+ primarily reflects red blood cell and leukocyte NAD+, which may not accurately represent tissue NAD+ in muscle, brain, or liver, where the clinically relevant depletion occurs.
Organic Acids Test (OAT): urine organic acids can suggest mitochondrial dysfunction downstream of NAD+ insufficiency; elevated lactate:pyruvate ratios, elevated alpha-ketoglutarate, and elevated citric acid cycle intermediates suggest impaired NADH oxidation.
Nutrient Zoomer (Vibrant Wellness): includes niacin/B3 status; since niacin is an NAD+ precursor, clinical B3 deficiency (pellagra at the extreme) will obviously impair NAD+ synthesis, though functional insufficiency short of clinical deficiency is harder to quantify.
Methylation Panel + Homocysteine: relevant because the kynurenine pathway and the NNMT methylation shunt both connect NAD+ metabolism to methylation; elevated homocysteine and low SAM-e can reflect excess methyl group consumption partly by NNMT.
Functional markers to track during NMN/NR supplementation:
- Fasting glucose and insulin (HOMA-IR)
- Inflammatory markers: hsCRP, IL-6, TNF-alpha
- Lipid panel
- Walking speed and grip strength (if sarcopenia is a concern)
- Energy and cognitive symptoms via symptom tracking
Mechanisms Of Action
Simple:
- NAD+ is like a rechargeable battery in every cell: it gets "discharged" to NADH when it picks up electrons from burning food, then gets "recharged" back to NAD+ by the electron transport chain so it can pick up more electrons; when this cycle slows down, so does every energy-producing process.
- NADPH is a different kind of battery used for protection and building: cells use it to neutralize free radicals (via the glutathione and thioredoxin systems), to synthesize fat, and to power immune cells when they kill bacteria; without enough NADPH, cells become fragile and vulnerable to oxidative damage.
- Sirtuins are molecular switches that activate when NAD+ is high (fasting, exercise) and go quiet when NAD+ is low (aging, inflammation, overeating); they deacetylate proteins to activate them, including the master mitochondrial regulator PGC-1alpha, making NAD+ a direct link between metabolic state and gene expression.
- When DNA gets damaged, PARP1 responds by consuming enormous amounts of NAD+ to signal for repair; in aging and chronic disease, so much DNA damage accumulates that PARP1 runs continuously, draining the NAD+ that sirtuins need to function.
- CD38 is the body's main NAD+ drain and gets turned up by aging and inflammation, which is why chronically inflamed people and older adults have lower NAD+ than young healthy people and why stopping inflammation is part of restoring NAD+ biology, not just adding precursors.
Advanced:
- The NAD+/NADH ratio as a metabolic rheostat and sirtuin regulator: The ratio of free NAD+ to free NADH in the cytoplasm (~700:1 in fasted state) and mitochondria (~10:1, more reduced) is a real-time readout of cellular energy status. Sirtuins have Km values for NAD+ in the range of 100-200 micromolar, which means they are operating well below saturating substrate concentrations under most physiological conditions. Small changes in free NAD+ concentration translate directly to changes in sirtuin catalytic rate. High-fat feeding and overnutrition reduce the NAD+/NADH ratio by flooding the TCA cycle with acetyl-CoA and generating NADH faster than Complex I can oxidize it, effectively silencing sirtuins via substrate competition even when total NAD+ appears adequate. This is why exercise and caloric restriction activate sirtuins not primarily by raising NAD+ but by shifting the ratio toward the oxidized form. R
- PARP1-SIRT1 competition and the futile cycle: PARP1 and SIRT1 both have high affinity for NAD+ (Km ~100-500 micromolar) and compete for the same substrate pool in the nucleus. PARP1 is activated within milliseconds of sensing a DNA single-strand break via its zinc finger domains, and can consume hundreds of NAD+ molecules per second during hyperactivation, generating massive poly-ADP-ribose chains. The resulting NAD+ depletion reduces SIRT1 activity in proportion, which reduces deacetylation of PARP2 (allowing PARP2 to suppress SIRT1 transcription), and reduces deacetylation of SIRT1-target transcription factors involved in mitochondrial biogenesis. Restoration of NAD+ (via NMN/NR or PARP inhibition) reverses this cascade, reactivating SIRT1 and improving mitochondrial function. This PARP-SIRT competition is directly measurable as increased SIRT1 activity following PARP1 inhibitor treatment in aging mouse tissues. R
- G6PD as the bidirectional NADPH switch for both oxidant generation and antioxidant defense: The same NADPH generated by G6PD from the PPP is substrate for both glutathione reductase (regenerating the antioxidant GSH) and NADPH oxidase (generating the oxidant superoxide). Both processes are simultaneously NADPH-dependent. In resting cells, most NADPH flux goes toward antioxidant maintenance. In activated neutrophils, G6PD is acutely activated (via protein translocation to the plasma membrane) and the respiratory burst redirects NADPH toward NOX2-mediated superoxide generation in the phagolysosome. In cancer cells, G6PD is upregulated by multiple oncogenic pathways (NRF2, mTOR, HIF-1alpha) to supply the large NADPH demand of rapidly proliferating cells for lipid biosynthesis and ROS defense against chemotherapy. IDH1 mutations in gliomas substitute the NADPH-generating IDH1 reaction with an NADPH-consuming reaction (alpha-KG + NADPH to 2-HG + NADP+), creating NADPH insufficiency, oxidative vulnerability, and accumulation of the oncometabolite 2-HG that inhibits alpha-KG-dependent dioxygenases (TET enzymes and histone demethylases), causing epigenetic reprogramming. R
More Research
- NAD+ levels decline 30-50% in human tissues between middle age and old age; this decline is mechanistically linked to elevated CD38 from inflammaging, increased PARP1 activation from accumulated DNA damage, reduced NAMPT expression from circadian disruption, and increased NNMT methylation shunting, all of which can theoretically be addressed. R
- Oral NMN and NR supplementation reliably raises blood NAD+ in humans across multiple RCTs; the most consistent functional benefits observed are in physical performance (walking speed, grip strength in older adults), vascular function (arterial stiffness), and metabolic markers (diastolic blood pressure, LDL) with variable effects on insulin sensitivity and no consistent effects on skeletal muscle mass. R R
- The biggest predictor of NMN/NR response appears to be baseline NAD+ level: individuals with NAD+ depletion (older adults, those with metabolic disease, chronic illness, or high inflammatory burden) show greater functional benefit from supplementation than those with already adequate NAD+ levels; this suggests targeting NAD+ precursor supplementation to populations with confirmed or likely depletion rather than using it preventively in young healthy individuals. R
- NR (2000 mg/day) showed significant improvement in cognition and fatigue in long-COVID patients over 24 weeks, a population with documented mitochondrial dysfunction and immune dysregulation where NAD+ depletion is biologically expected. R
- G6PD deficiency, the most common human enzyme deficiency (400 million affected worldwide), is a clinically important constraint on NADPH biology that is rarely considered in supplement recommendations; oxidizing supplements including high-dose vitamin C, certain antioxidants, and antimalarials can precipitate hemolytic crises in G6PD-deficient individuals whose NADPH is already compromised. R
- PARP inhibition (FDA-approved drugs olaparib, niraparib, rucaparib for BRCA-deficient cancers) is a pharmaceutical application of NAD+ biology; the synthetic lethality in BRCA-mutant tumors is directly downstream of NAD+ depletion from PARP hyperactivation in cells that cannot repair the resulting double-strand breaks. R
- CD38 inhibition via apigenin and quercetin represents a complementary and mechanistically distinct strategy from NAD+ precursor supplementation; addressing both sides of the NAD+ equation (supply via NMN/NR and demand via CD38/PARP inhibition) is more complete than either approach alone; apigenin at 50-100mg and quercetin at 500-1000mg are reasonable additions alongside NMN or NR for those specifically targeting NAD+ optimization.
Jacob Gordon
INHC, FMT-C
Board Certified Health Coach
I spent years battling unexplained chronic illness before discovering biohacking, epigenetics, and functional medicine. Now I share that research at MyBioHack to help others find their own answers.
Book a ConsultationRelated Protocols & Supplements
Deep-dive chapters and recommended supplements for this topic
Quercetin
500mg 2x/day
Vitamin D3 + K2
5000 IU + 200mcg/day
Magnesium Glycinate
400mg at bedtime